

Integrating Hi-C links with assembly graphs for chromosome-scale assembly
- PMID: 31433799
- PMCID: PMC6719893
- DOI: 10.1371/journal.pcbi.1007273
Integrating Hi-C links with assembly graphs for chromosome-scale assembly
Abstract
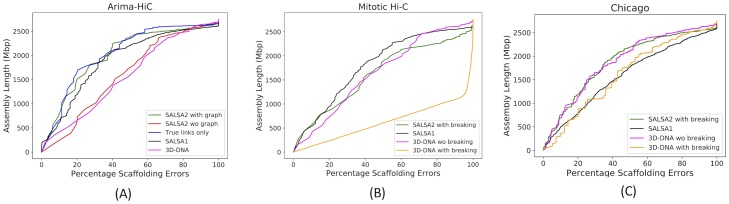
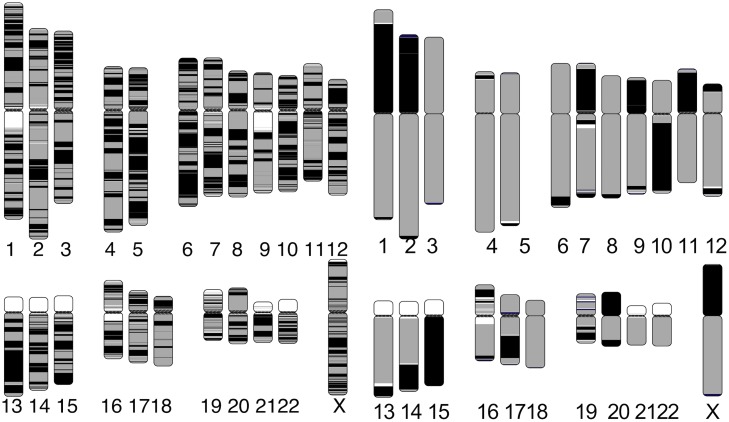
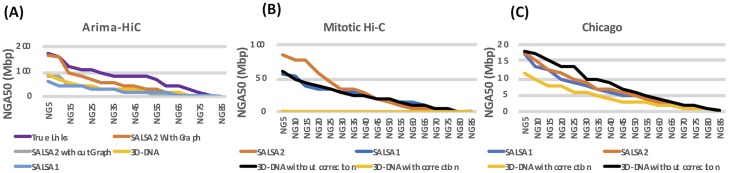
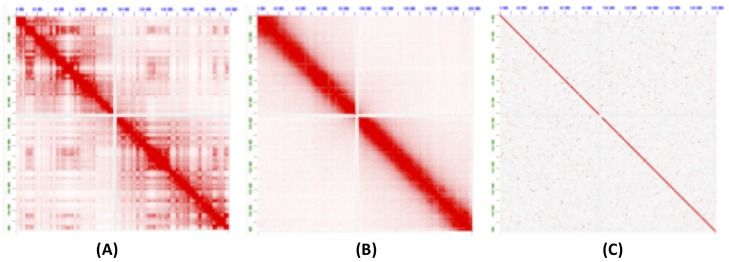
Long-read sequencing and novel long-range assays have revolutionized de novo genome assembly by automating the reconstruction of reference-quality genomes. In particular, Hi-C sequencing is becoming an economical method for generating chromosome-scale scaffolds. Despite its increasing popularity, there are limited open-source tools available. Errors, particularly inversions and fusions across chromosomes, remain higher than alternate scaffolding technologies. We present a novel open-source Hi-C scaffolder that does not require an a priori estimate of chromosome number and minimizes errors by scaffolding with the assistance of an assembly graph. We demonstrate higher accuracy than the state-of-the-art methods across a variety of Hi-C library preparations and input assembly sizes. The Python and C++ code for our method is openly available at https://github.com/machinegun/SALSA.
Conflict of interest statement
Sergey Koren has received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. Anthony Schmitt and Siddarth Selvaraj are employees of Arima Genomics, a company commercializing Hi-C DNA sequencing technologies.
Figures
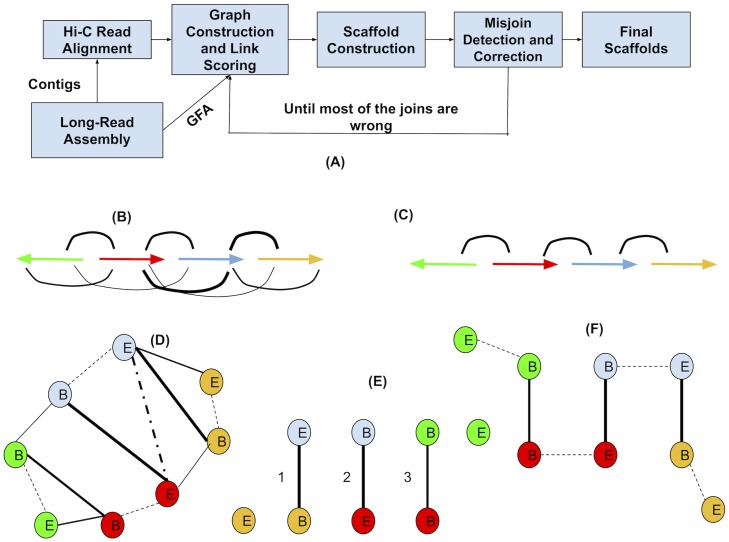
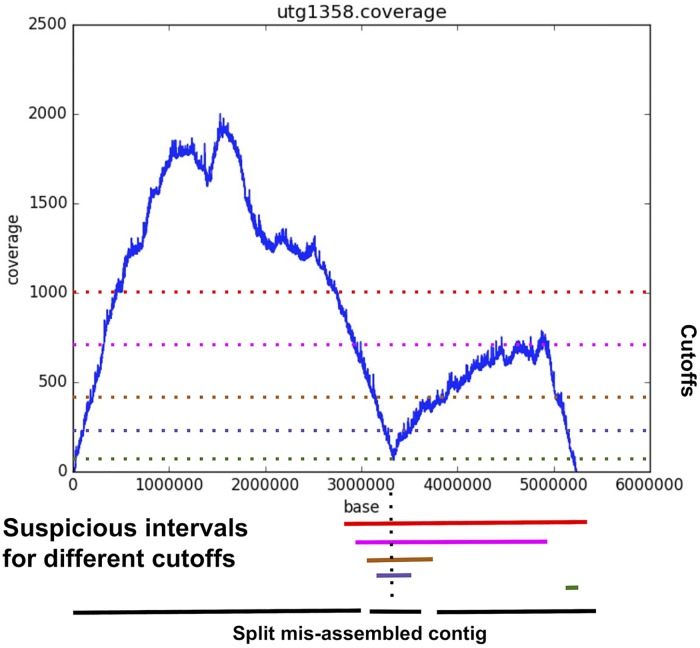
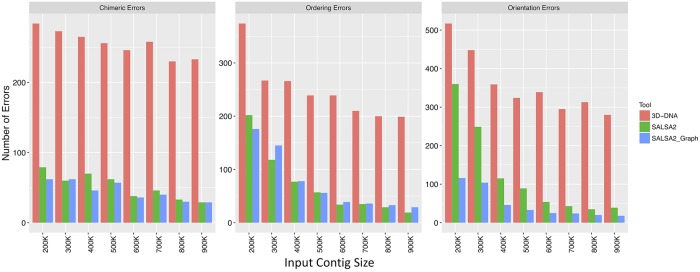
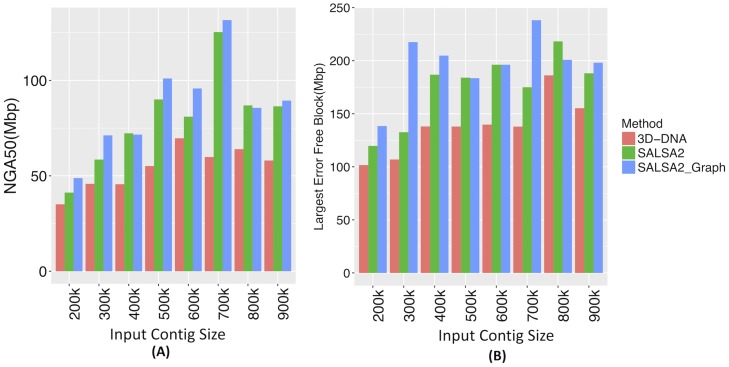
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
