

Genotoxic stress-triggered β-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis
- PMID: 31434880
- PMCID: PMC6704105
- DOI: 10.1038/s41467-019-11696-7
Genotoxic stress-triggered β-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis
Abstract
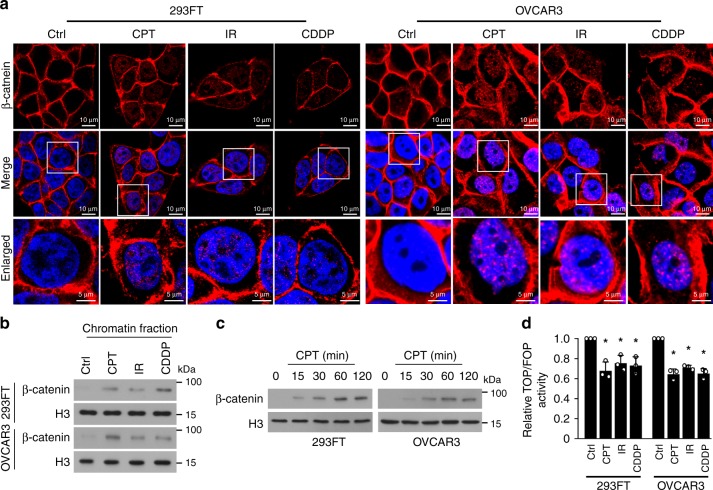
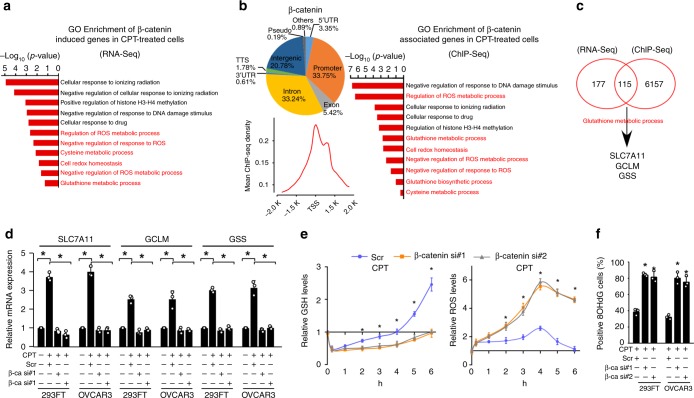
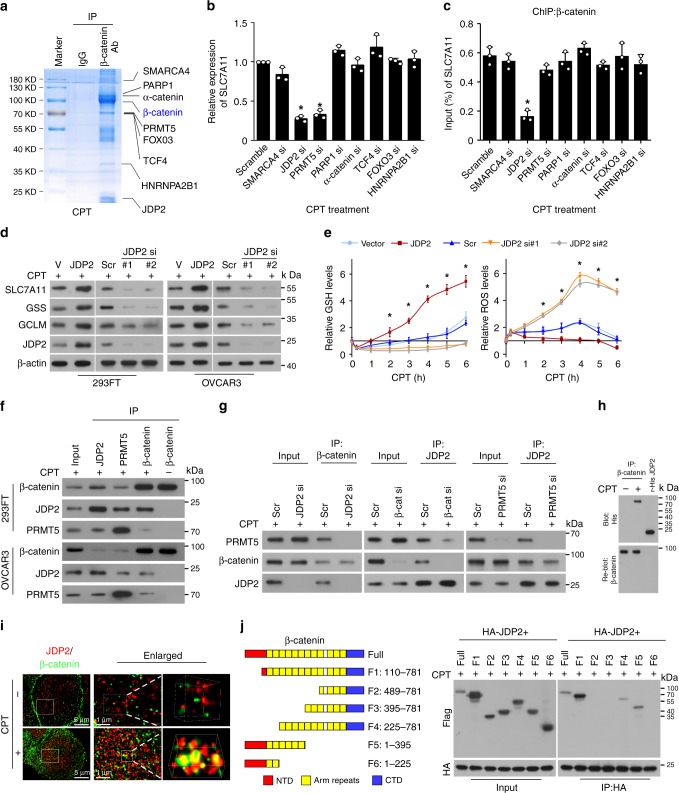
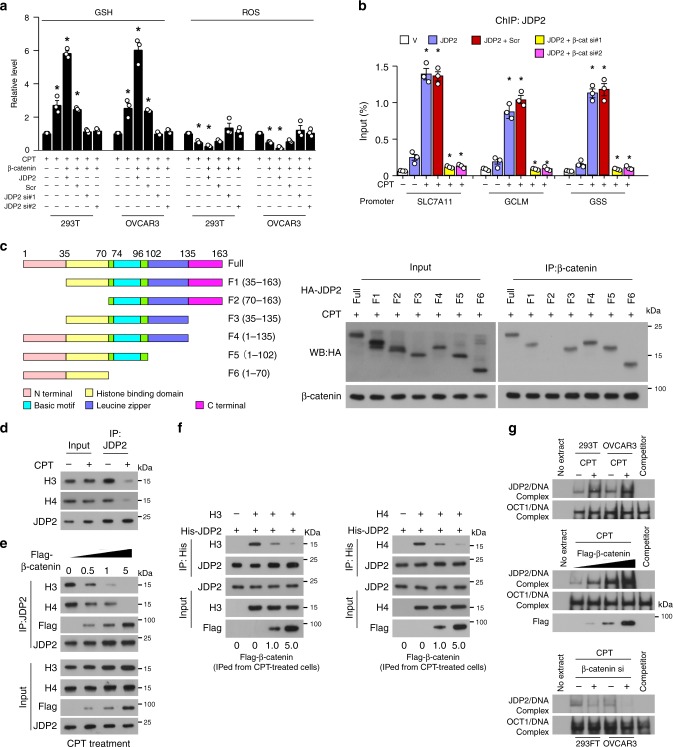
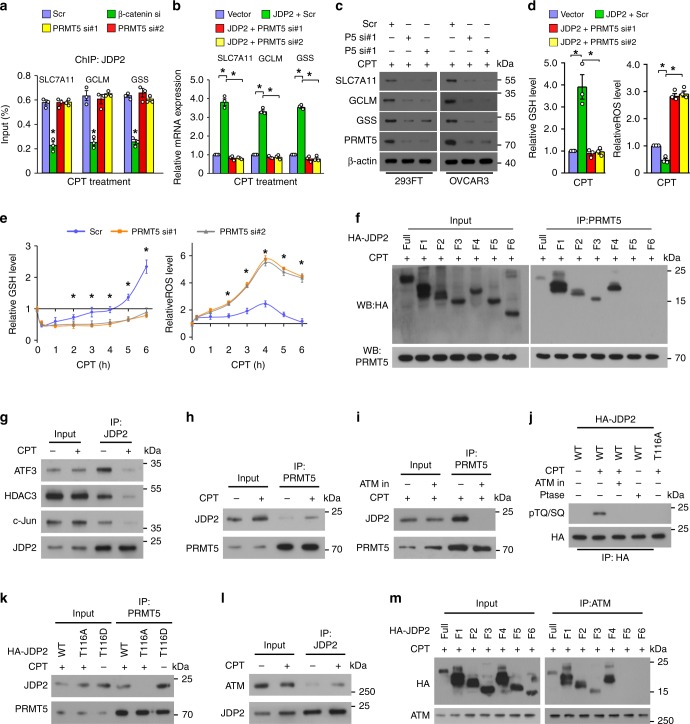
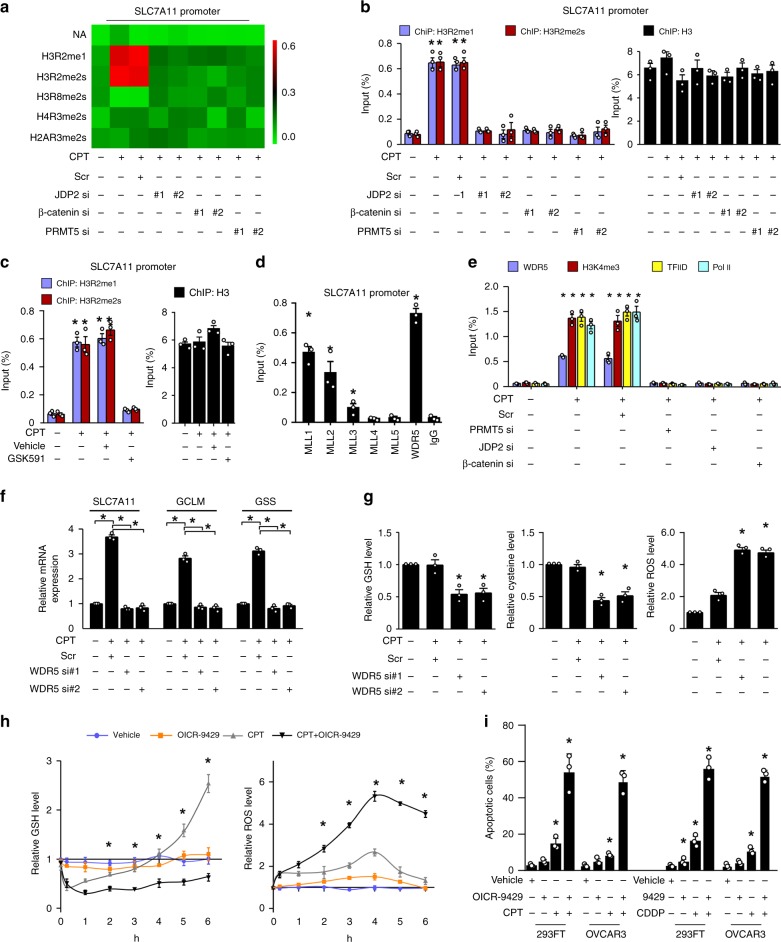
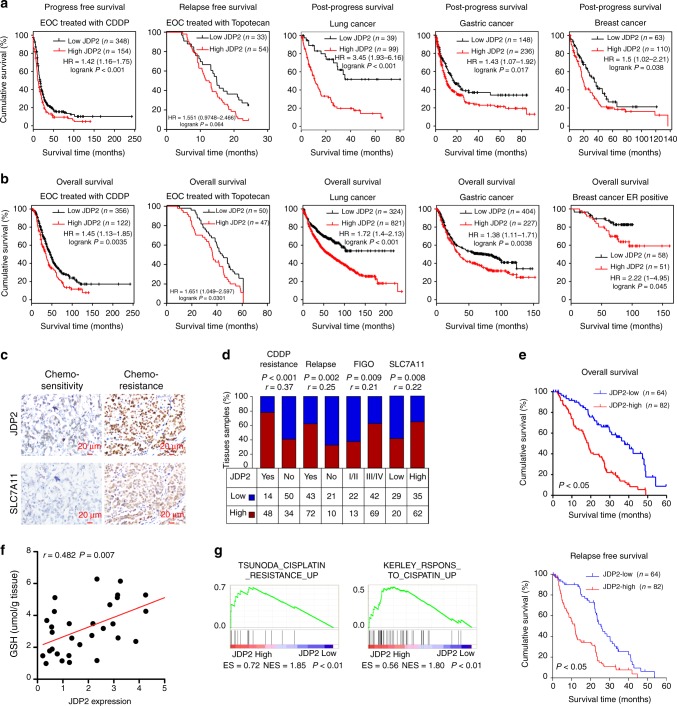
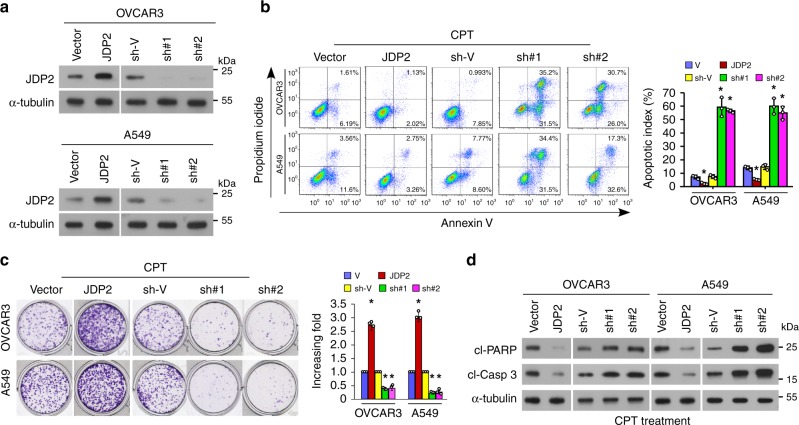
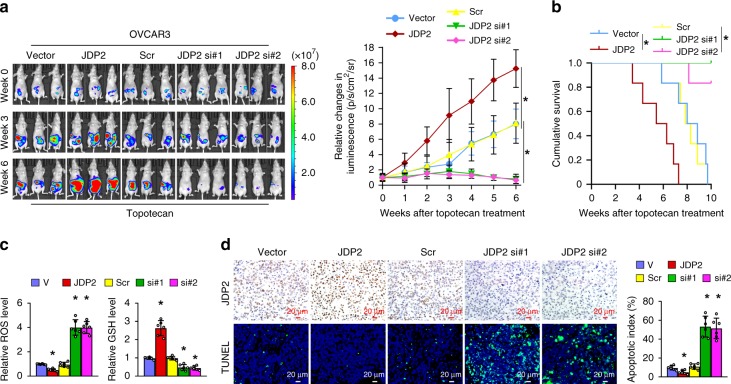
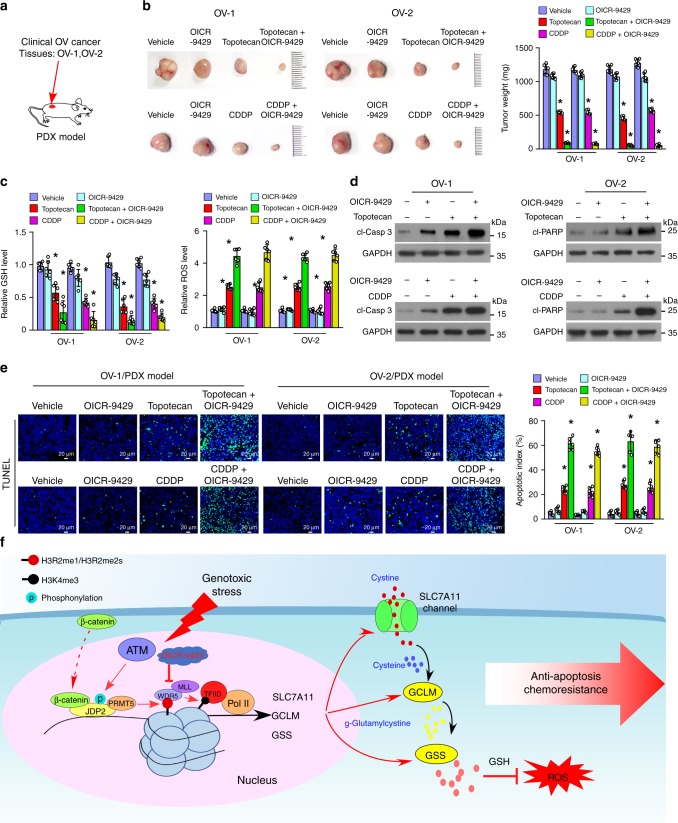
The mechanisms underlying how cells subjected to genotoxic stress reestablish reduction-oxidation (redox) homeostasis to scavenge genotoxic stress-induced reactive oxygen species (ROS), which maintains the physiological function of cellular processes and cell survival, remain unclear. Herein, we report that, via a TCF-independent mechanism, genotoxic stress induces the enrichment of β-catenin in chromatin, where it forms a complex with ATM phosphorylated-JDP2 and PRMT5. This elicits histone H3R2me1/H3R2me2s-induced transcriptional activation by the recruitment of the WDR5/MLL methyltransferase complexes and concomitant H3K4 methylation at the promoters of multiple genes in GSH-metabolic cascade. Treatment with OICR-9429, a small-molecule antagonist of the WDR5-MLL interaction, inhibits the β-catenin/JDP2/PRMT5 complex-reestablished GSH metabolism, leading to a lethal increase in the already-elevated levels of ROS in the genotoxic-agent treated cancer cells. Therefore, our results unveil a plausible role for β-catenin in reestablishing redox homeostasis upon genotoxic stress and shed light on the mechanisms of inducible chemotherapy resistance in cancer.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
