

Anthocyanins and Their Metabolites as Therapeutic Agents for Neurodegenerative Disease
- PMID: 31443476
- PMCID: PMC6770078
- DOI: 10.3390/antiox8090333
Anthocyanins and Their Metabolites as Therapeutic Agents for Neurodegenerative Disease
Abstract
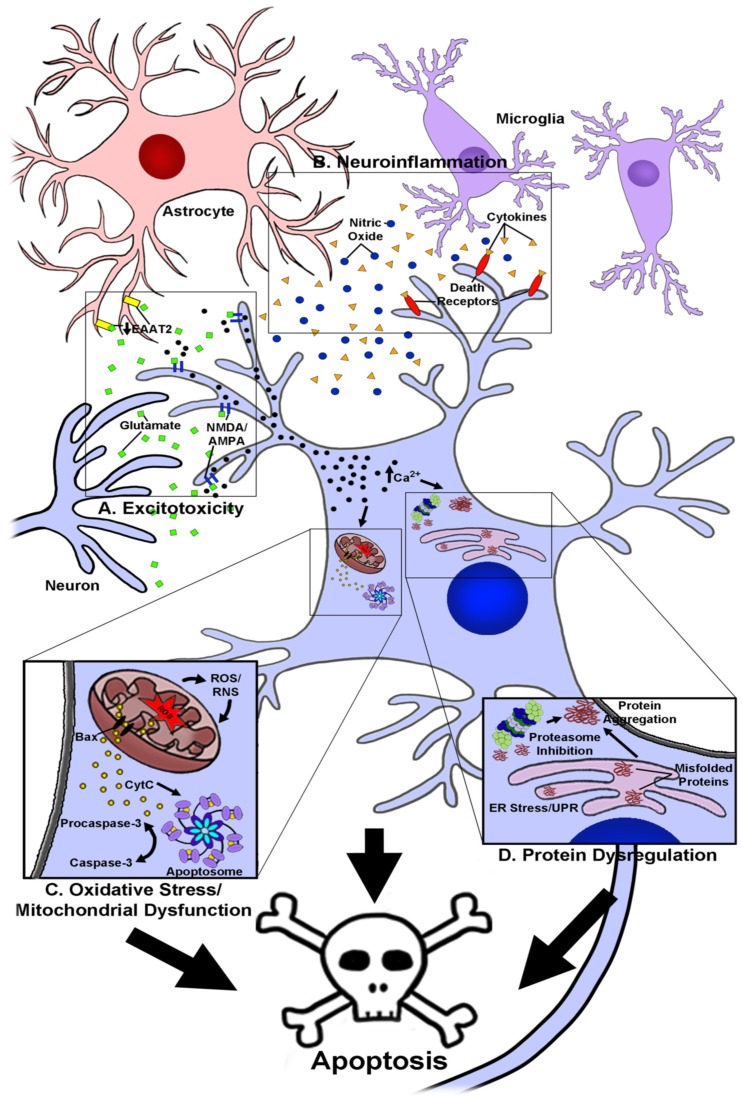
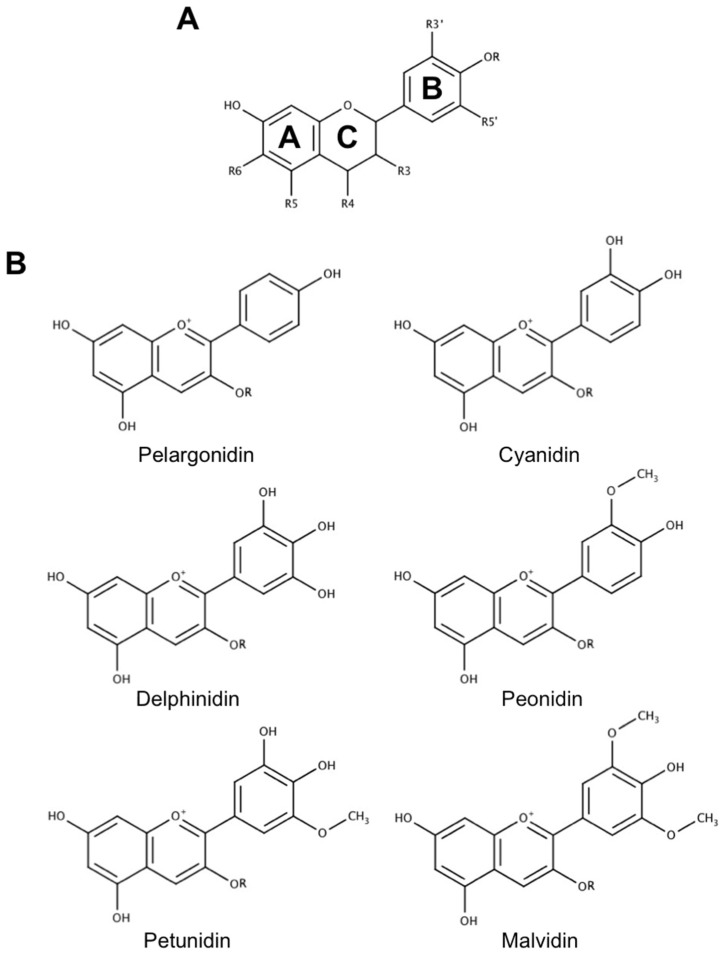
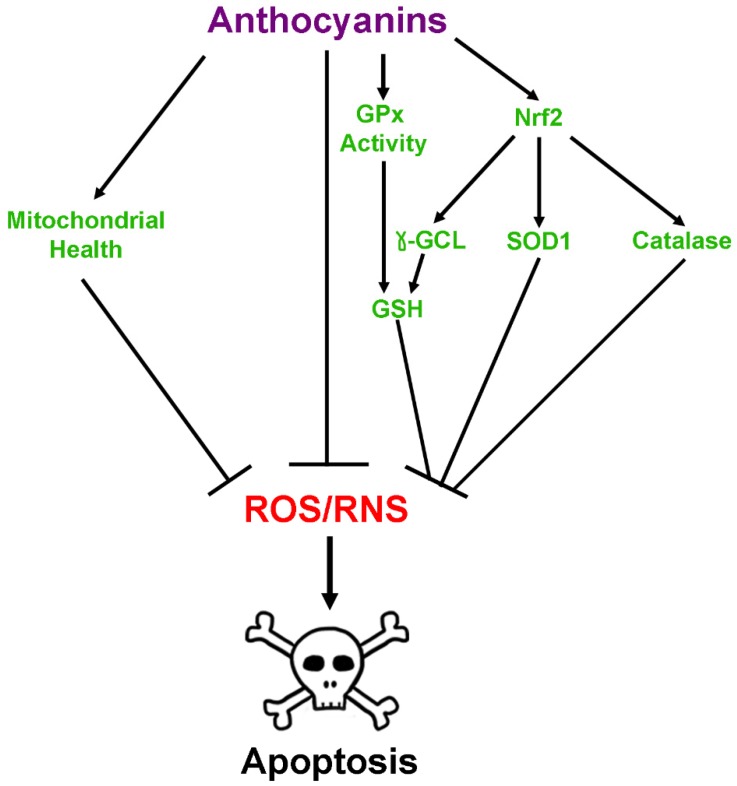
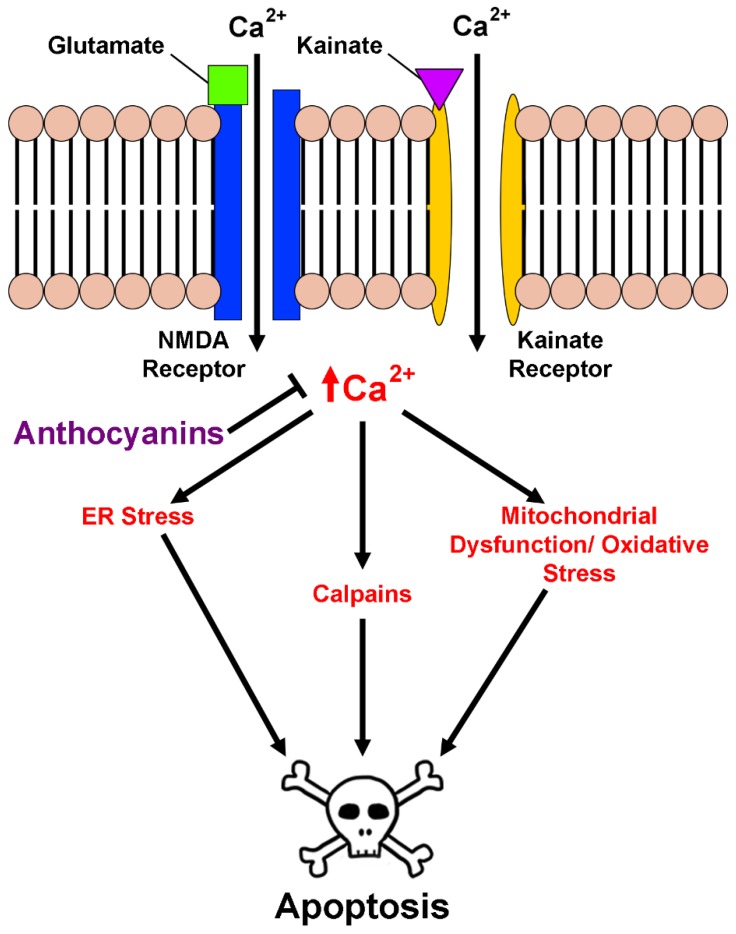
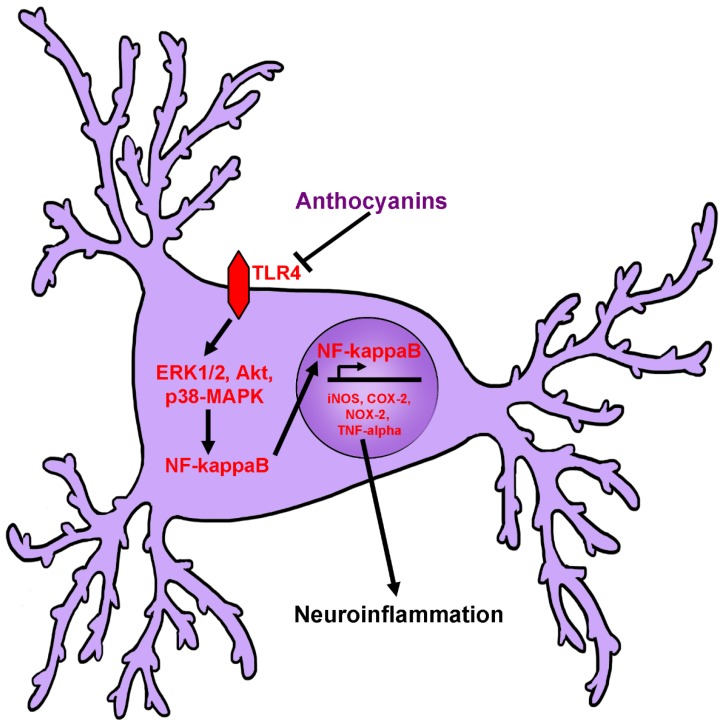
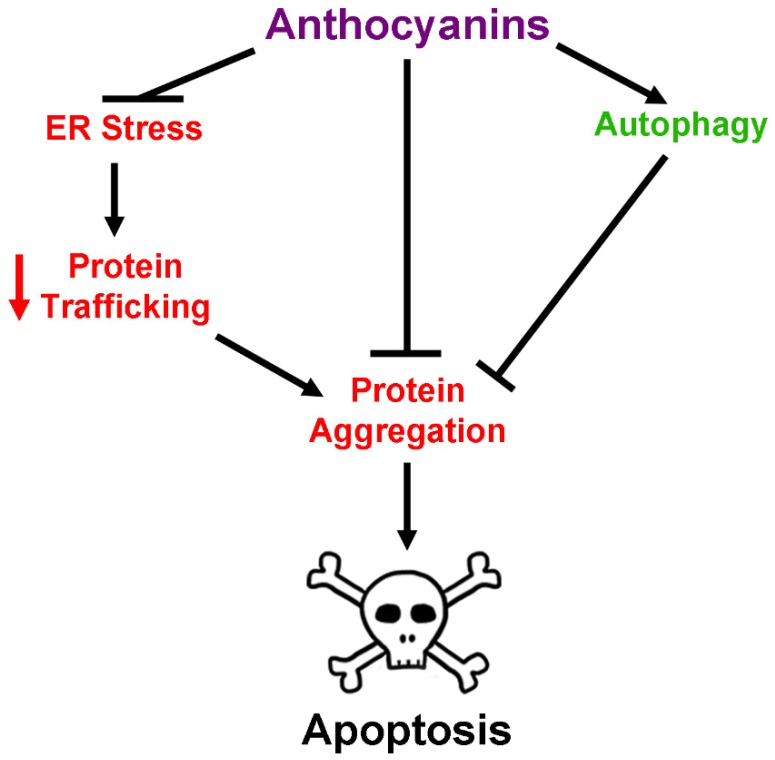
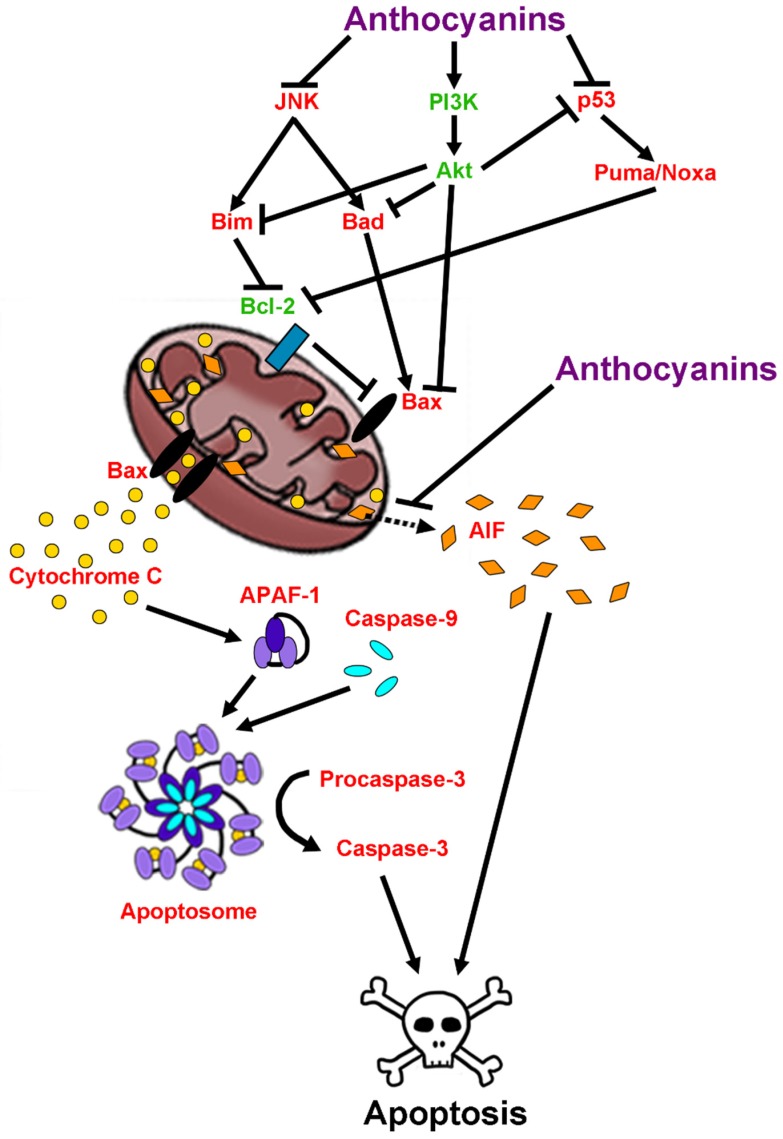
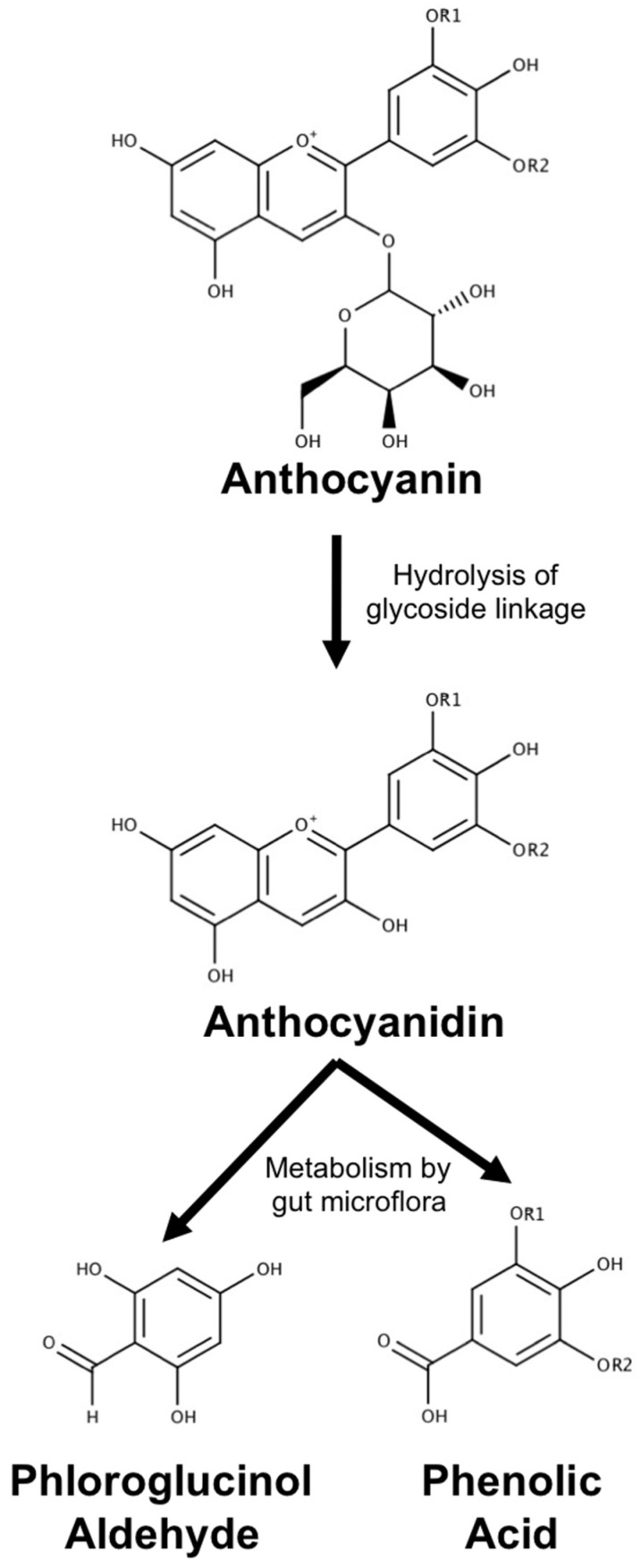
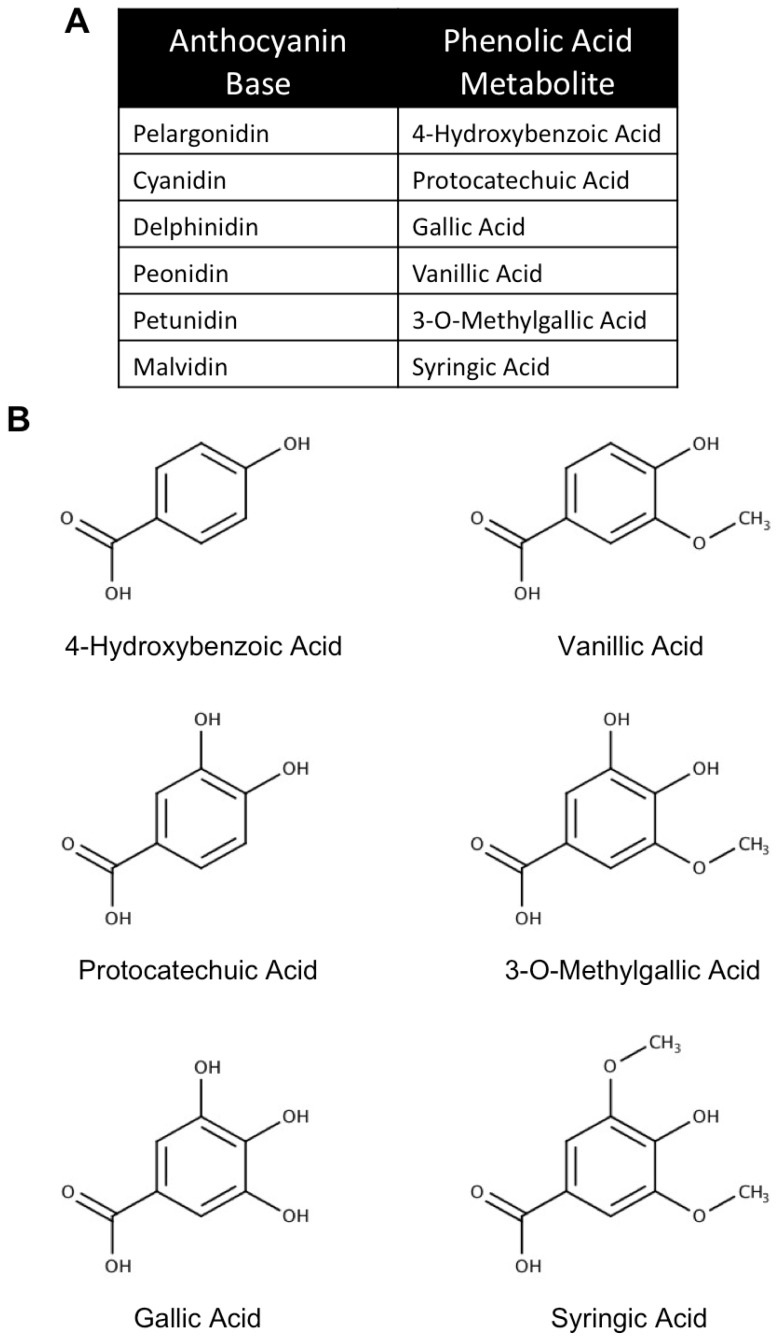
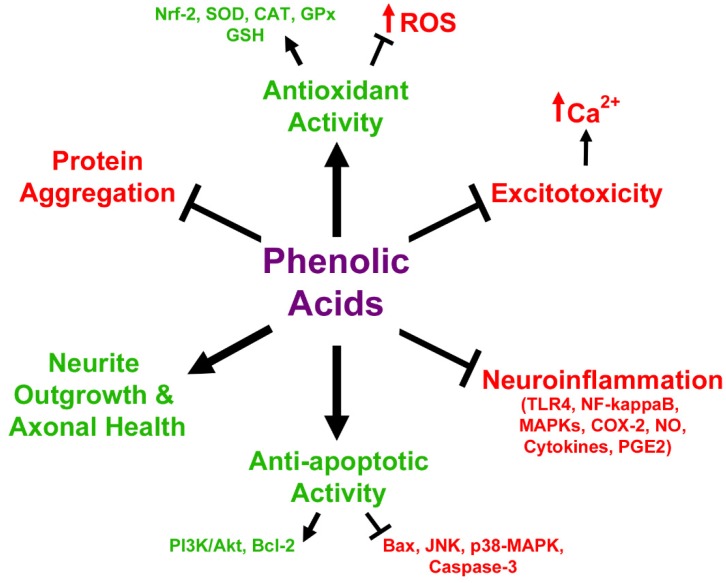
Neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS), are characterized by the death of neurons within specific regions of the brain or spinal cord. While the etiology of many neurodegenerative diseases remains elusive, several factors are thought to contribute to the neurodegenerative process, such as oxidative and nitrosative stress, excitotoxicity, endoplasmic reticulum stress, protein aggregation, and neuroinflammation. These processes culminate in the death of vulnerable neuronal populations, which manifests symptomatically as cognitive and/or motor impairments. Until recently, most treatments for these disorders have targeted single aspects of disease pathology; however, this strategy has proved largely ineffective, and focus has now turned towards therapeutics which target multiple aspects underlying neurodegeneration. Anthocyanins are unique flavonoid compounds that have been shown to modulate several of the factors contributing to neuronal death, and interest in their use as therapeutics for neurodegeneration has grown in recent years. Additionally, due to observations that the bioavailability of anthocyanins is low relative to that of their metabolites, it has been proposed that anthocyanin metabolites may play a significant part in mediating the beneficial effects of an anthocyanin-rich diet. Thus, in this review, we will explore the evidence evaluating the neuroprotective and therapeutic potential of anthocyanins and their common metabolites for treating neurodegenerative diseases.
Keywords: Alzheimer’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; anthocyanins; flavonoids; inflammation; neurodegeneration; neuroprotection; oxidative stress; phenolic acids.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
