Genomic landscape and chronological reconstruction of driver events in multiple myeloma
- PMID: 31444325
- PMCID: PMC6707220
- DOI: 10.1038/s41467-019-11680-1
Genomic landscape and chronological reconstruction of driver events in multiple myeloma
Abstract
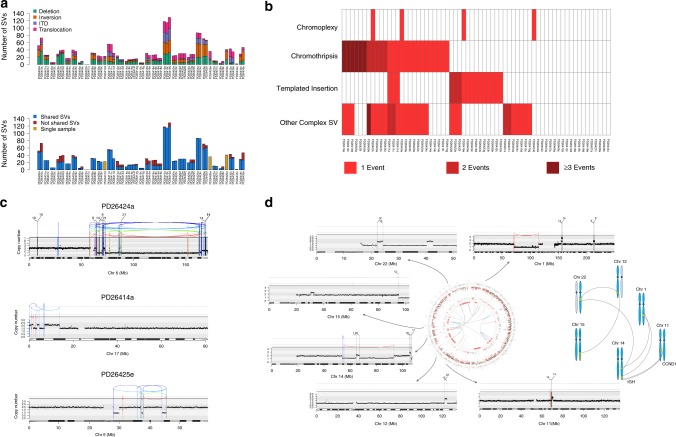
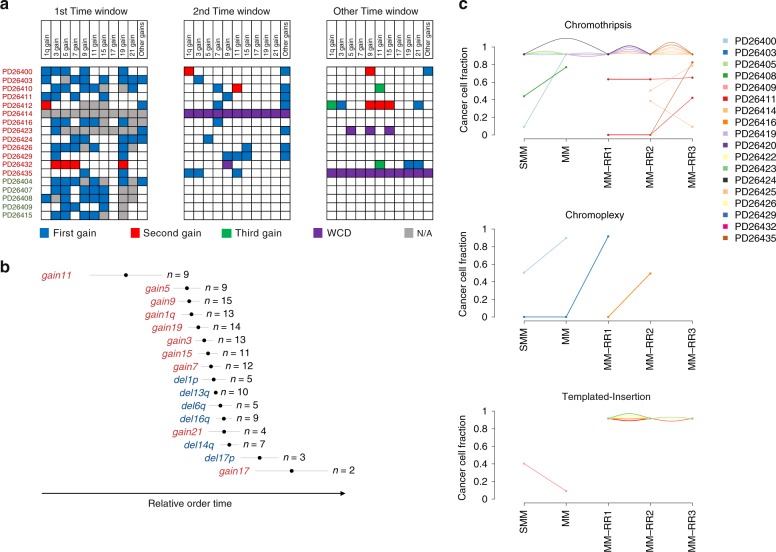
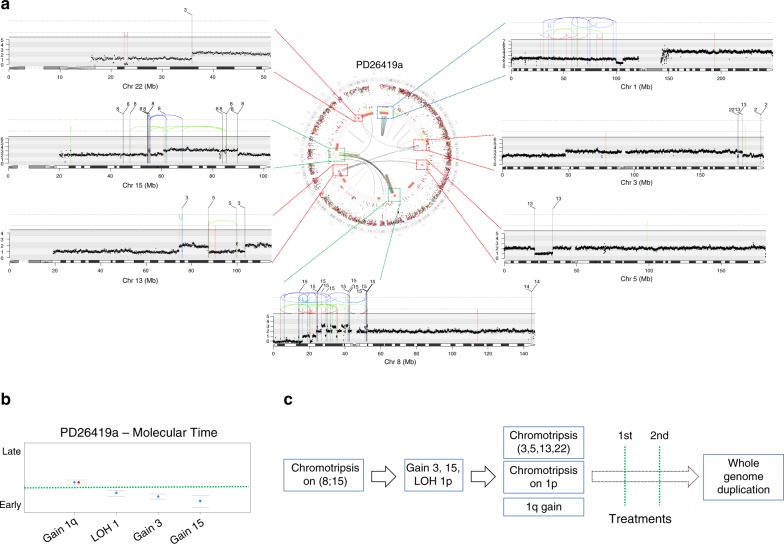
The multiple myeloma (MM) genome is heterogeneous and evolves through preclinical and post-diagnosis phases. Here we report a catalog and hierarchy of driver lesions using sequences from 67 MM genomes serially collected from 30 patients together with public exome datasets. Bayesian clustering defines at least 7 genomic subgroups with distinct sets of co-operating events. Focusing on whole genome sequencing data, complex structural events emerge as major drivers, including chromothripsis and a novel replication-based mechanism of templated insertions, which typically occur early. Hyperdiploidy also occurs early, with individual trisomies often acquired in different chronological windows during evolution, and with a preferred order of acquisition. Conversely, positively selected point mutations, whole genome duplication and chromoplexy events occur in later disease phases. Thus, initiating driver events, drawn from a limited repertoire of structural and numerical chromosomal changes, shape preferred trajectories of evolution that are biologically relevant but heterogeneous across patients.
Conflict of interest statement
The authors declare no competing interests.
Figures