

Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived Friedreich's ataxia cardiomyocytes
- PMID: 31446150
- PMCID: PMC6853280
- DOI: 10.1016/j.scr.2019.101529
Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived Friedreich's ataxia cardiomyocytes
Abstract
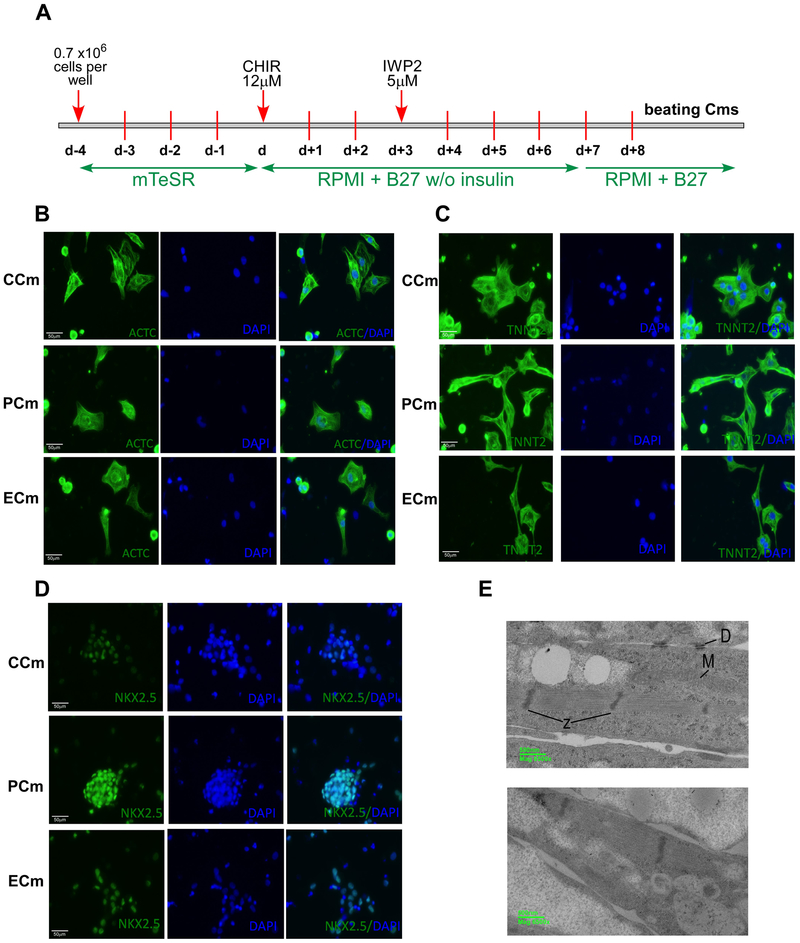
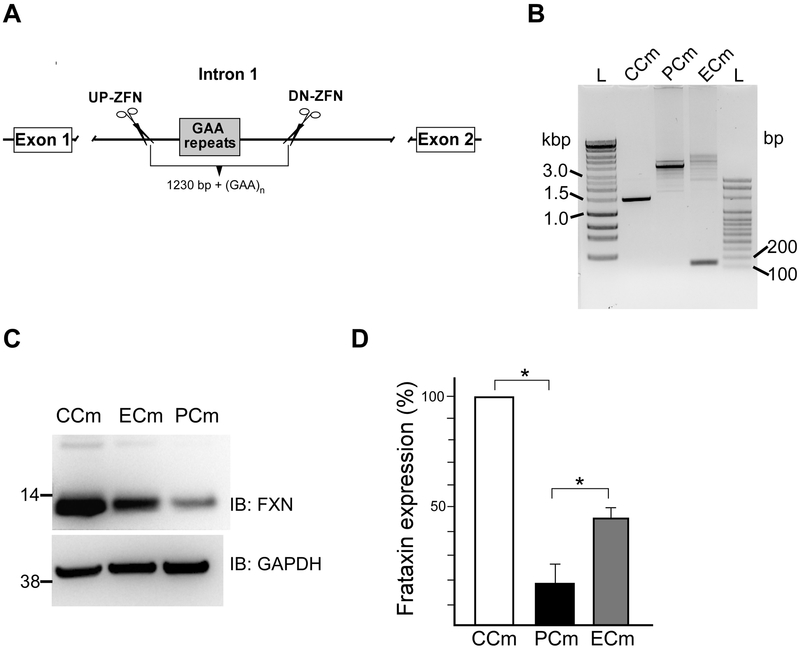
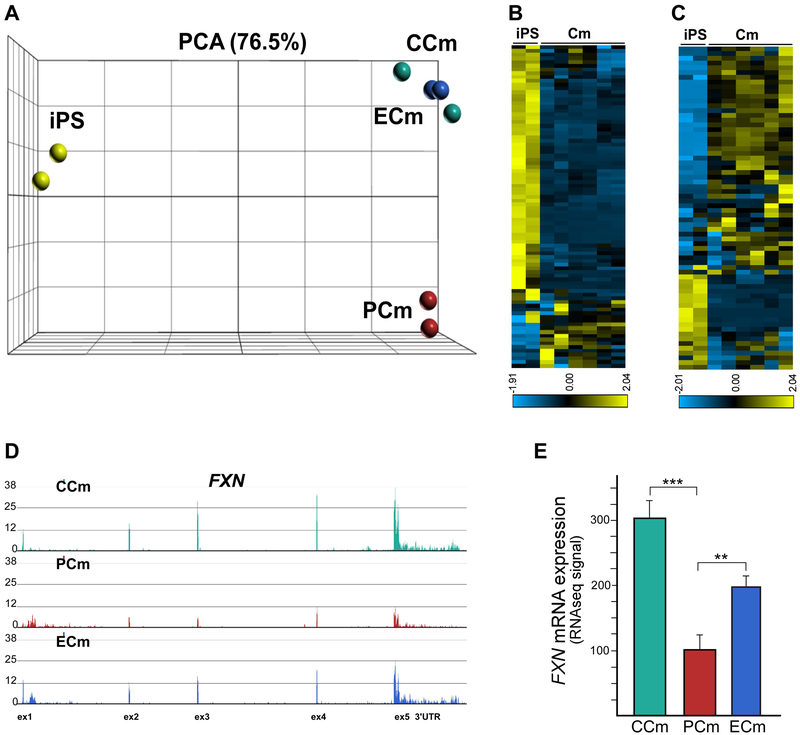
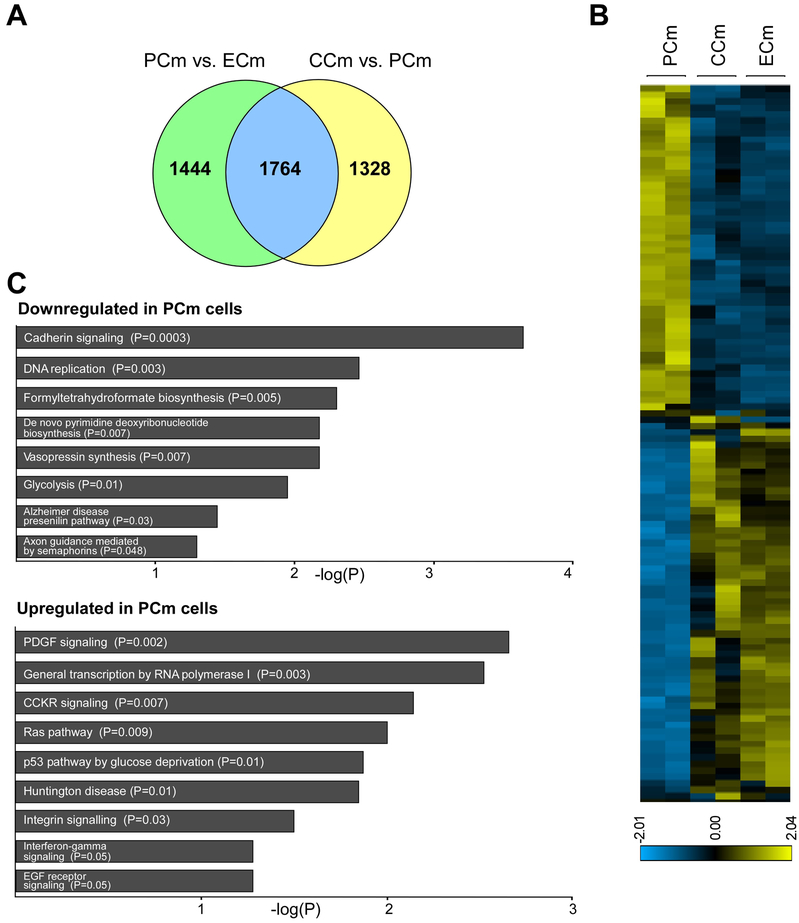
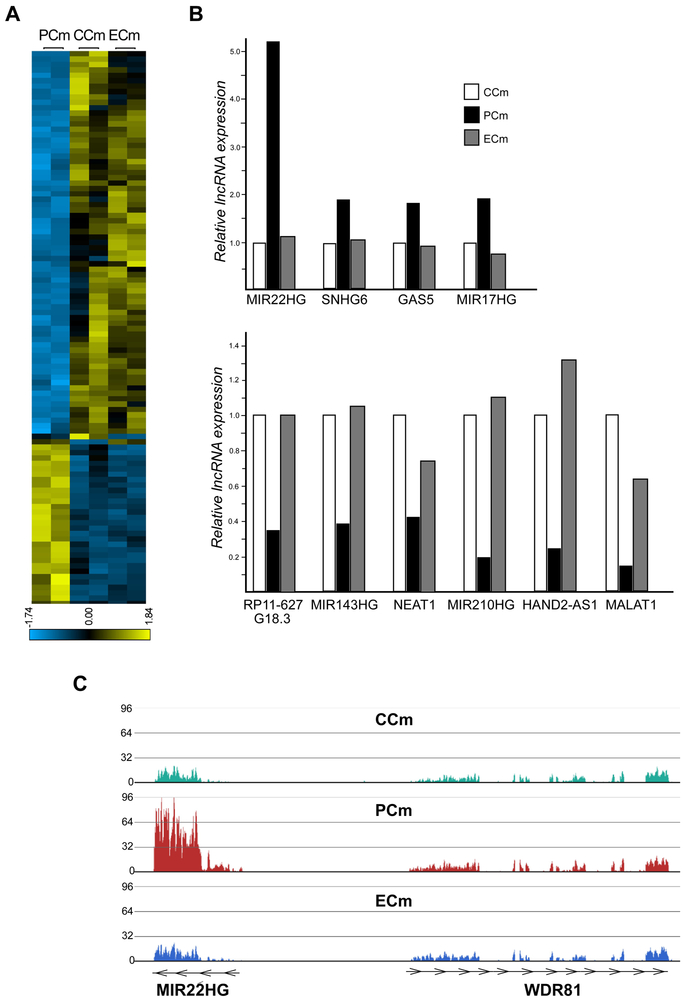
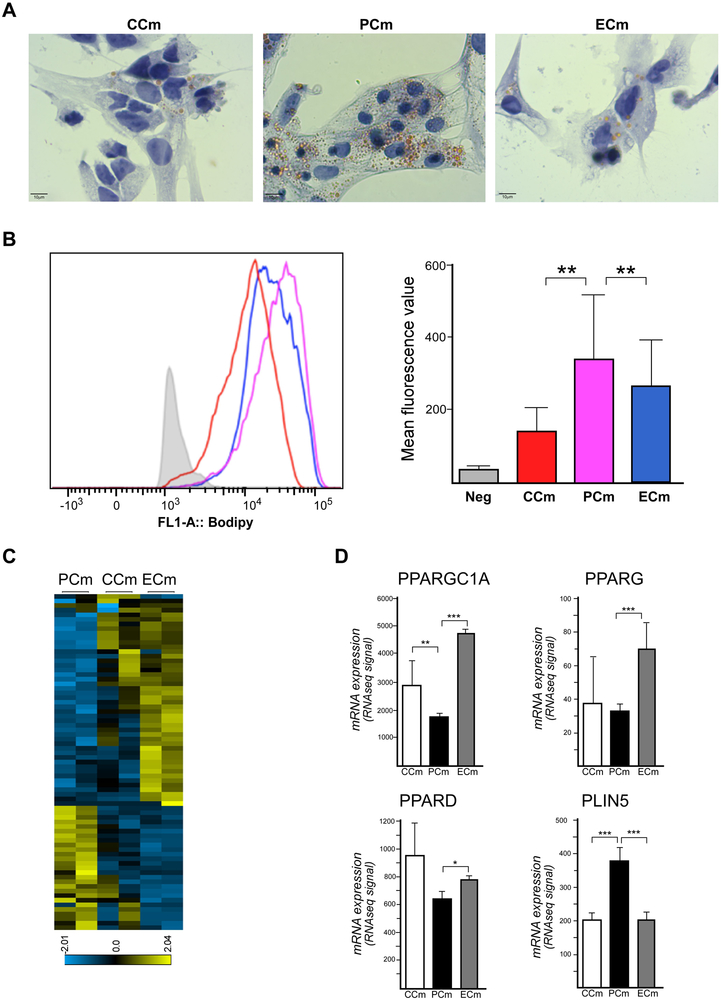
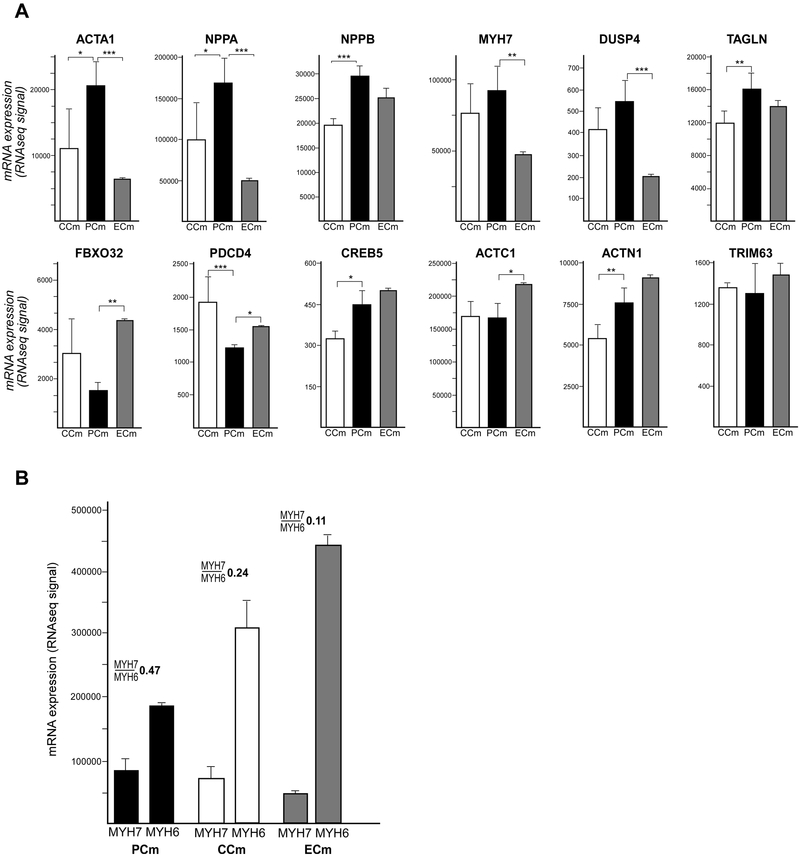
Friedreich's ataxia is caused by large homozygous, intronic expansions of GAA repeats in the frataxin (FXN) gene, resulting in severe downregulation of its expression. Pathogenic repeats are located in intron one, hence patients express unaffected FXN protein, albeit in low quantities. Although FRDA symptoms typically afflict the nervous system, hypertrophic cardiomyopathy is the predominant cause of death. Our studies were conducted using cardiomyocytes differentiated from induced pluripotent stem cells derived from control individuals, FRDA patients, and isogenic cells corrected by zinc finger nucleases-mediated excision of pathogenic expanded GAA repeats. This correction of the FXN gene removed the primary trigger of the transcription defect, upregulated frataxin expression, reduced pathological lipid accumulation observed in patient cardiomyocytes, and reversed gene expression signatures of FRDA cardiomyocytes. Transcriptome analyses revealed hypertrophy-specific expression signatures unique to FRDA cardiomyocytes, and emphasized similarities between unaffected and ZFN-corrected FRDA cardiomyocytes. Thus, the iPSC-derived FRDA cardiomyocytes exhibit various molecular defects characteristic for cellular models of cardiomyopathy that can be corrected by genome editing of the expanded GAA repeats. These results underscore the utility of genome editing in generating isogenic cellular models of FRDA and the potential of this approach as a future therapy for this disease.
Keywords: Cardiomyocytes; Friedreich's ataxia; GAA repeats; Genome editing; Isogenic iPSC; Lipid metabolism.
Copyright © 2019 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests
None
Figures
References
-
- Bulteau AL, O'Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, and Szweda LI, 2004. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305, 242–245. - PubMed
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, De Donato S, Mandel JL, Cocozza S, Koenig M, and Pandolfo M, 1996. Friedreich's ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
