

Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water
- PMID: 31448259
- PMCID: PMC6691143
- DOI: 10.3389/fchem.2019.00540
Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water
Abstract
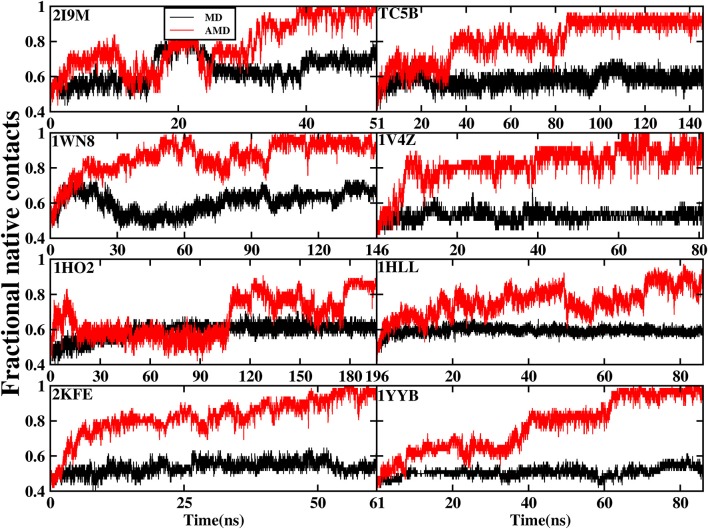
In this study, we examined the folding processes of eight helical proteins (2I9M, TC5B, 1WN8, 1V4Z, 1HO2, 1HLL, 2KFE, and 1YYB) at room temperature using the explicit solvent model under the AMBER14SB force field with the accelerated molecular dynamics (AMD) and traditional molecular dynamics (MD), respectively. We analyzed and compared the simulation results obtained by these two methods based on several aspects, such as root mean square deviation (RMSD), native contacts, cluster analysis, folding snapshots, free energy landscape, and the evolution of the radius of gyration, which showed that these eight proteins were successfully and consistently folded into the corresponding native structures by AMD simulations carried out at room temperature. In addition, the folding occurred in the range of 40~180 ns after starting from the linear structures of the eight proteins at 300 K. By contrast, these stable folding structures were not found when the traditional molecular dynamics (MD) simulation was used. At the same time, the influence of high temperatures (350, 400, and 450 K) is also further investigated. Study found that the simulation efficiency of AMD is higher than that of MD simulations, regardless of the temperature. Of these temperatures, 300 K is the most suitable temperature for protein folding for all systems. To further investigate the efficiency of AMD, another trajectory was simulated for eight proteins with the same linear structure but different random seeds at 300 K. Both AMD trajectories reached the correct folded structures. Our result clearly shows that AMD simulation are a highly efficient and reliable method for the study of protein folding.
Keywords: accelerated molecular dynamics simulation; explicit water; free energy landscape; helical protein; protein folding.
Figures











References
LinkOut - more resources
Full Text Sources
Miscellaneous