

Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network
- PMID: 31448325
- PMCID: PMC6688864
- DOI: 10.1126/sciadv.aav6490
Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network
Abstract
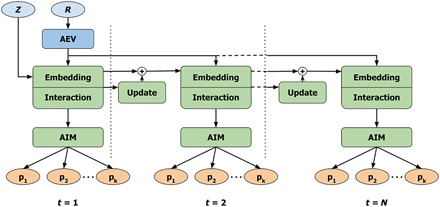
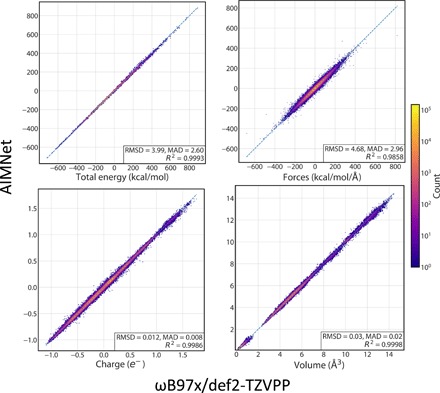
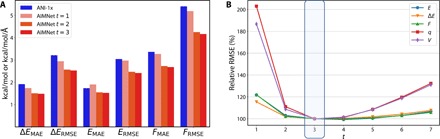
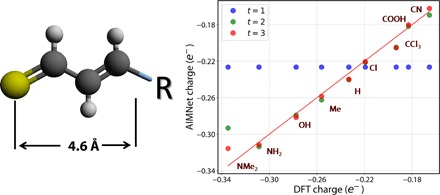
Atomic and molecular properties could be evaluated from the fundamental Schrodinger's equation and therefore represent different modalities of the same quantum phenomena. Here, we present AIMNet, a modular and chemically inspired deep neural network potential. We used AIMNet with multitarget training to learn multiple modalities of the state of the atom in a molecular system. The resulting model shows on several benchmark datasets state-of-the-art accuracy, comparable to the results of orders of magnitude more expensive DFT methods. It can simultaneously predict several atomic and molecular properties without an increase in the computational cost. With AIMNet, we show a new dimension of transferability: the ability to learn new targets using multimodal information from previous training. The model can learn implicit solvation energy (SMD method) using only a fraction of the original training data and an archive median absolute deviation error of 1.1 kcal/mol compared to experimental solvation free energies in the MNSol database.
Figures
References
-
- Rupp M., Tkatchenko A., Müller K.-R., von Lilienfeld O. A., Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
