

Extremely Fast and Accurate Open Modification Spectral Library Searching of High-Resolution Mass Spectra Using Feature Hashing and Graphics Processing Units
- PMID: 31448616
- PMCID: PMC6886738
- DOI: 10.1021/acs.jproteome.9b00291
Extremely Fast and Accurate Open Modification Spectral Library Searching of High-Resolution Mass Spectra Using Feature Hashing and Graphics Processing Units
Abstract
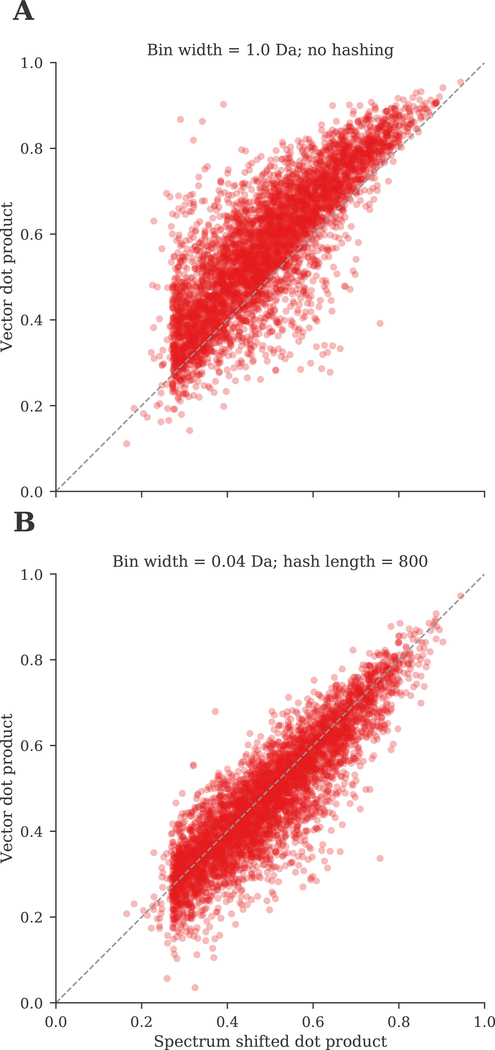
Open modification searching (OMS) is a powerful search strategy to identify peptides with any type of modification. OMS works by using a very wide precursor mass window to allow modified spectra to match against their unmodified variants, after which the modification types can be inferred from the corresponding precursor mass differences. A disadvantage of this strategy, however, is the large computational cost, because each query spectrum has to be compared against a multitude of candidate peptides. We have previously introduced the ANN-SoLo tool for fast and accurate open spectral library searching. ANN-SoLo uses approximate nearest neighbor indexing to speed up OMS by selecting only a limited number of the most relevant library spectra to compare to an unknown query spectrum. Here we demonstrate how this candidate selection procedure can be further optimized using graphics processing units. Additionally, we introduce a feature hashing scheme to convert high-resolution spectra to low-dimensional vectors. On the basis of these algorithmic advances, along with low-level code optimizations, the new version of ANN-SoLo is up to an order of magnitude faster than its initial version. This makes it possible to efficiently perform open searches on a large scale to gain a deeper understanding about the protein modification landscape. We demonstrate the computational efficiency and identification performance of ANN-SoLo based on a large data set of the draft human proteome. ANN-SoLo is implemented in Python and C++. It is freely available under the Apache 2.0 license at https://github.com/bittremieux/ANN-SoLo .
Keywords: approximate nearest neighbor indexing; feature hashing; graphics processing unit; mass spectrometry; open modification searching; post-translational modification; proteomics; spectral library.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
