

Amikacin liposome inhalation suspension for chronic Pseudomonas aeruginosa infection in cystic fibrosis
- PMID: 31451351
- PMCID: PMC9078215
- DOI: 10.1016/j.jcf.2019.08.001
Amikacin liposome inhalation suspension for chronic Pseudomonas aeruginosa infection in cystic fibrosis
Abstract
Background: Shortcomings of inhaled antibiotic treatments for Pseudomonas aeruginosa infection in patients with cystic fibrosis (CF) include poor drug penetration, inactivation by sputum, poor efficiency due to protective biofilm, and short residence in the lung.
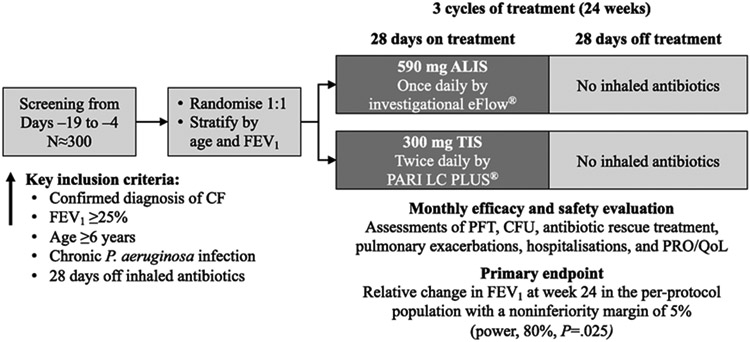
Methods: Eligible patients with forced expiratory volume in 1 s (FEV1) ≥25% of predicted value at screening and CF with chronic P. aeruginosa infection were randomly assigned to receive 3 treatment cycles (28 days on, 28 days off) of amikacin liposome inhalation suspension (ALIS, 590 mg QD) or tobramycin inhalation solution (TIS, 300 mg BID). The primary endpoint was noninferiority of ALIS vs TIS in change from baseline to day 168 in FEV1 (per-protocol population). Secondary endpoints included change in respiratory symptoms by Cystic Fibrosis Questionnaire-Revised (CFQ-R).
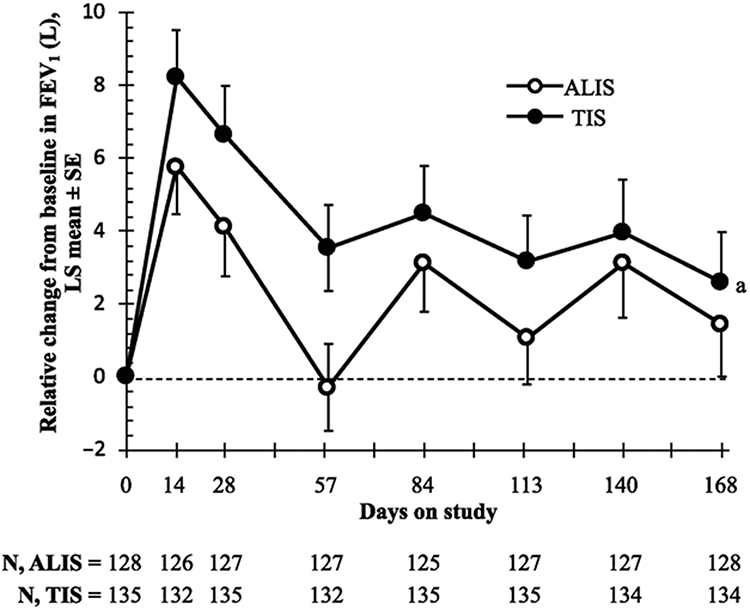
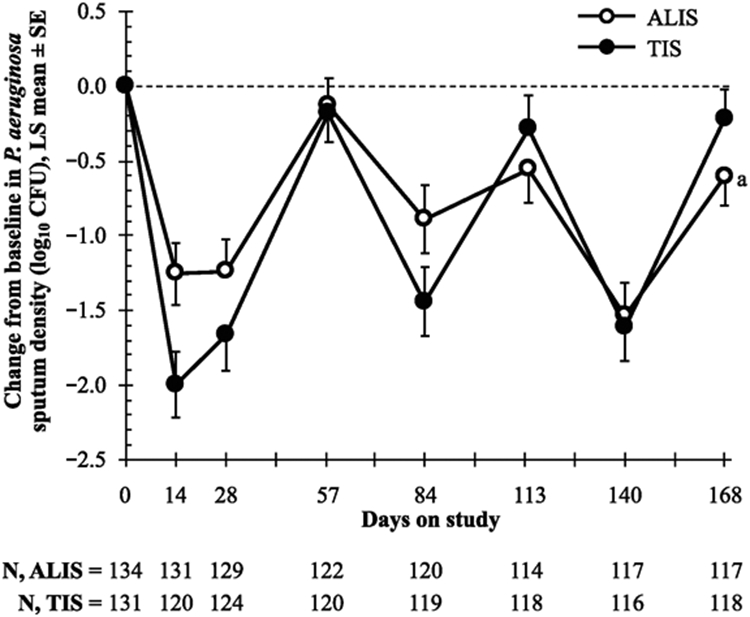
Results: The study was conducted February 2012 to September 2013. ALIS was noninferior to TIS (95% CI, -4.95 to 2.34) for relative change in FEV1 (L) from baseline. The mean increases in CFQ-R score from baseline on the Respiratory Symptoms scale suggested clinically meaningful improvement in both arms at the end of treatment in cycle 1 and in the ALIS arm at the end of treatment in cycles 2 and 3; however, the changes were not statistically significant between the 2 treatment arms. Treatment-emergent adverse events (TEAEs) were reported in most patients (ALIS, 84.5%; TIS, 78.8%). Serious TEAEs occurred in 17.6% and 19.9% of patients, respectively; most were hospitalisations for infective pulmonary exacerbation of CF.
Conclusions: Cyclical dosing of once-daily ALIS was noninferior to cyclical twice-daily TIS in improving lung function. ClinicalTrials.gov Identifier: NCT01315678.
Keywords: ALIS; Amikacin liposome inhalation suspension; CFQ-R; Cystic fibrosis; LAI; Liposomal amikacin for inhalation; Pseudomonas aeruginosa.
Copyright © 2019 European Cystic Fibrosis Society. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Cystic Fibrosis Foundation. Patient Registry 2012. Annual Data Report, www.cysticfibrosisdata.org/LiteratureRetrieve.aspx?ID=149756; 2012 [accessed 12 December 2018].
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
