

Cell Death and Neurodegeneration
- PMID: 31451511
- PMCID: PMC6996453
- DOI: 10.1101/cshperspect.a036434
Cell Death and Neurodegeneration
Abstract
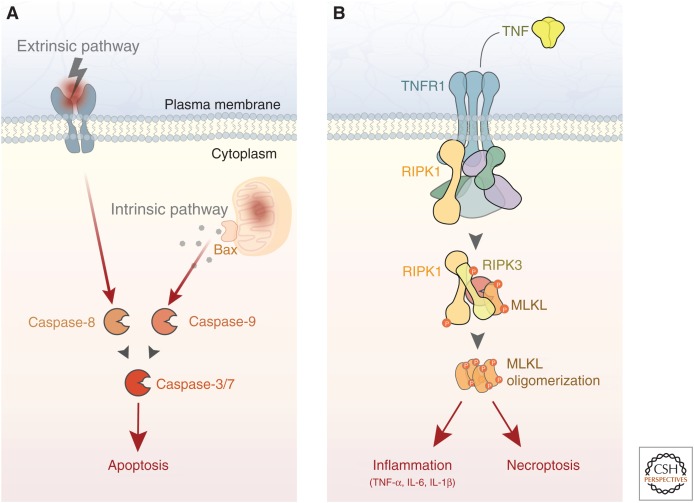
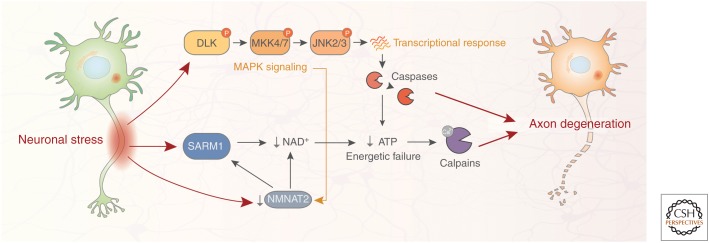
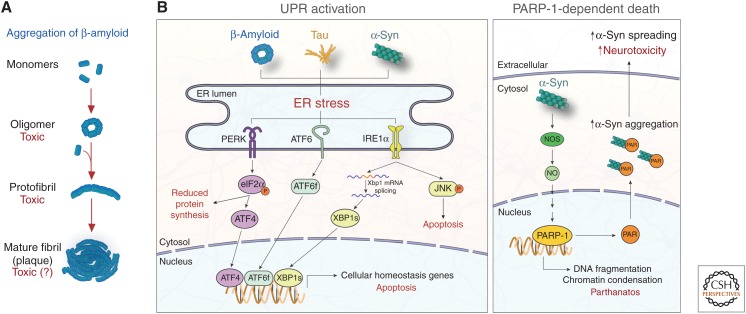
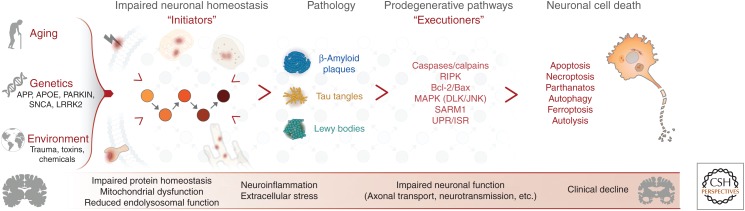
Neurodegenerative disease is characterized by the progressive deterioration of neuronal function caused by the degeneration of synapses, axons, and ultimately the death of nerve cells. An increased understanding of the mechanisms underlying altered cellular homeostasis and neurodegeneration is critical to the development of effective treatments for disease. Here, we review what is known about neuronal cell death and how it relates to our understanding of neurodegenerative disease pathology. First, we discuss prominent molecular signaling pathways that drive neuronal loss, and highlight the upstream cell biology underlying their activation. We then address how neuronal death may occur during disease in response to neuron intrinsic and extrinsic stressors. An improved understanding of the molecular mechanisms underlying neuronal dysfunction and cell death will open up avenues for clinical intervention in a field lacking disease-modifying treatments.
Copyright © 2020 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC III, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, et al. 2013. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci 33: 9498–9507. 10.1523/jneurosci.5397-12.2013 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical