

Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction
- PMID: 31451640
- PMCID: PMC6744920
- DOI: 10.1073/pnas.1910574116
Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction
Erratum in
-
Correction for Qian et al., Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction.Proc Natl Acad Sci U S A. 2025 Mar 11;122(10):e2501913122. doi: 10.1073/pnas.2501913122. Epub 2025 Feb 20. Proc Natl Acad Sci U S A. 2025. PMID: 39977333 Free PMC article. No abstract available.
Abstract
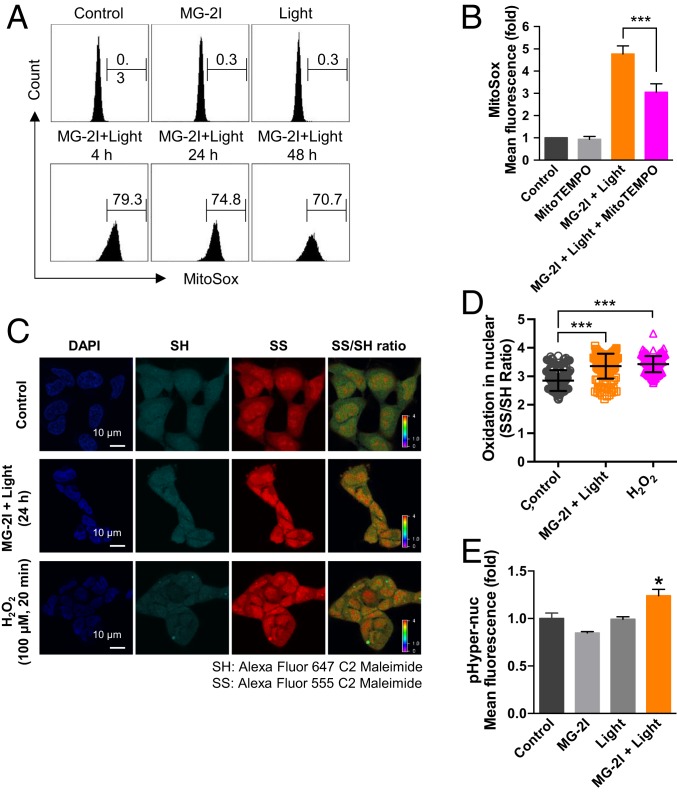
Reactive oxygen species (ROS) play important roles in aging, inflammation, and cancer. Mitochondria are an important source of ROS; however, the spatiotemporal ROS events underlying oxidative cellular damage from dysfunctional mitochondria remain unresolved. To this end, we have developed and validated a chemoptogenetic approach that uses a mitochondrially targeted fluorogen-activating peptide (Mito-FAP) to deliver a photosensitizer MG-2I dye exclusively to this organelle. Light-mediated activation (660 nm) of the Mito-FAP-MG-2I complex led to a rapid loss of mitochondrial respiration, decreased electron transport chain complex activity, and mitochondrial fragmentation. Importantly, one round of singlet oxygen produced a persistent secondary wave of mitochondrial superoxide and hydrogen peroxide lasting for over 48 h after the initial insult. By following ROS intermediates, we were able to detect hydrogen peroxide in the nucleus through ratiometric analysis of the oxidation of nuclear cysteine residues. Despite mitochondrial DNA (mtDNA) damage and nuclear oxidative stress induced by dysfunctional mitochondria, there was a lack of gross nuclear DNA strand breaks and apoptosis. Targeted telomere analysis revealed fragile telomeres and telomere loss as well as 53BP1-positive telomere dysfunction-induced foci (TIFs), indicating that DNA double-strand breaks occurred exclusively in telomeres as a direct consequence of mitochondrial dysfunction. These telomere defects activated ataxia-telangiectasia mutated (ATM)-mediated DNA damage repair signaling. Furthermore, ATM inhibition exacerbated the Mito-FAP-induced mitochondrial dysfunction and sensitized cells to apoptotic cell death. This profound sensitivity of telomeres through hydrogen peroxide induced by dysregulated mitochondria reveals a crucial mechanism of telomere-mitochondria communication underlying the pathophysiological role of mitochondrial ROS in human diseases.
Keywords: ATM signaling; DNA damage response; mitochondria; singlet oxygen; telomere.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: M.P.B. is a founder of Sharp Edge Labs, a company applying the FAP-fluorogen technology.
Figures






References
-
- Van Houten B., Woshner V., Santos J. H., Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst.) 5, 145–152 (2006). - PubMed
-
- de Moura M. B., dos Santos L. S., Van Houten B., Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ. Mol. Mutagen. 51, 391–405 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
