

Distinct Tissue-Specific Roles for the Disease-Associated Autophagy Genes ATG16L2 and ATG16L1
- PMID: 31451676
- PMCID: PMC6761021
- DOI: 10.4049/jimmunol.1800419
Distinct Tissue-Specific Roles for the Disease-Associated Autophagy Genes ATG16L2 and ATG16L1
Abstract
The clear role of autophagy in human inflammatory diseases such as Crohn disease was first identified by genome-wide association studies and subsequently dissected in multiple mechanistic studies. ATG16L1 has been particularly well studied in knockout and hypomorph settings as well as models recapitulating the Crohn disease-associated T300A polymorphism. Interestingly, ATG16L1 has a single homolog, ATG16L2, which is independently implicated in diseases, including Crohn disease and systemic lupus erythematosus. However, the contribution of ATG16L2 to canonical autophagy pathways and other cellular functions is poorly understood. To better understand its role, we generated and analyzed the first, to our knowledge, ATG16L2 knockout mouse. Our results show that ATG16L1 and ATG16L2 contribute very distinctly to autophagy and cellular ontogeny in myeloid, lymphoid, and epithelial lineages. Dysregulation of any of these lineages could contribute to complex diseases like Crohn disease and systemic lupus erythematosus, highlighting the value of examining cell-specific effects. We also identify a novel genetic interaction between ATG16L2 and epithelial ATG16L1. These findings are discussed in the context of how these genes may contribute distinctly to human disease.
Copyright © 2019 by The American Association of Immunologists, Inc.
Conflict of interest statement
Figures
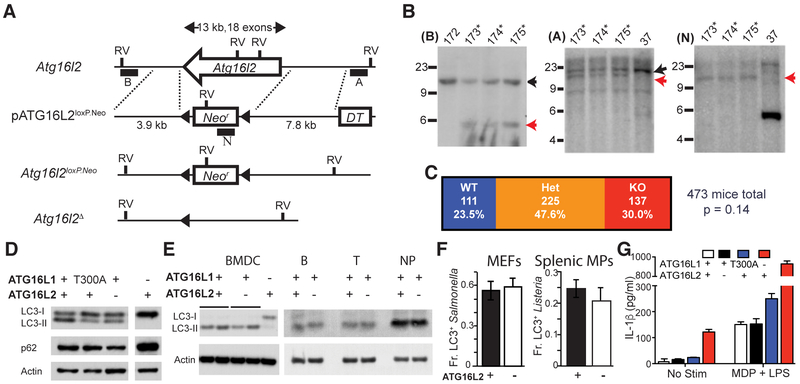

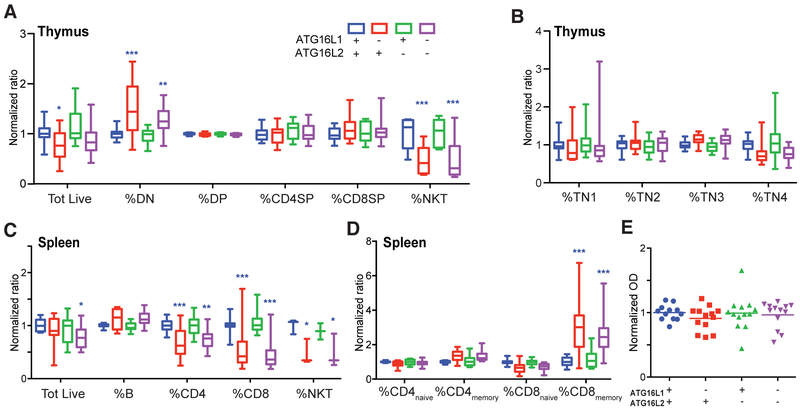

References
-
- Mizushima N, and Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147: 728–741. - PubMed
-
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, NIDDK IBD Genetics Consortium, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, van Gossum A, Zelenika D, Franchimont D, Hugot J-P, de Vos M, Vermeire S, Louis E, Belgian-French IBD Consortium, Wellcome Trust Case Control Consortium, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, and Daly MJ. 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature Genetics 40: 955–962. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
