

Uncovering the biosynthetic potential of rare metagenomic DNA using co-occurrence network analysis of targeted sequences
- PMID: 31451725
- PMCID: PMC6710260
- DOI: 10.1038/s41467-019-11658-z
Uncovering the biosynthetic potential of rare metagenomic DNA using co-occurrence network analysis of targeted sequences
Abstract
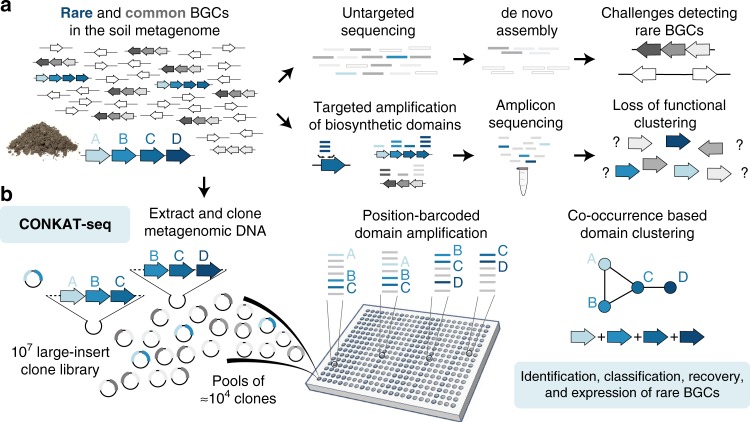
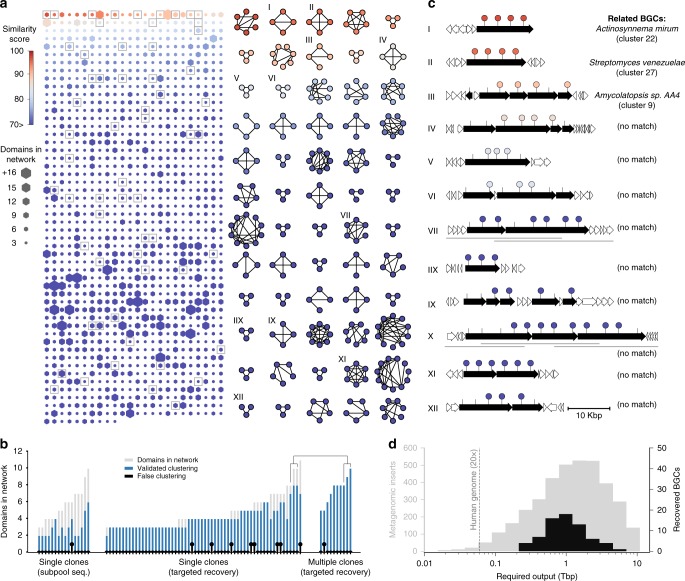
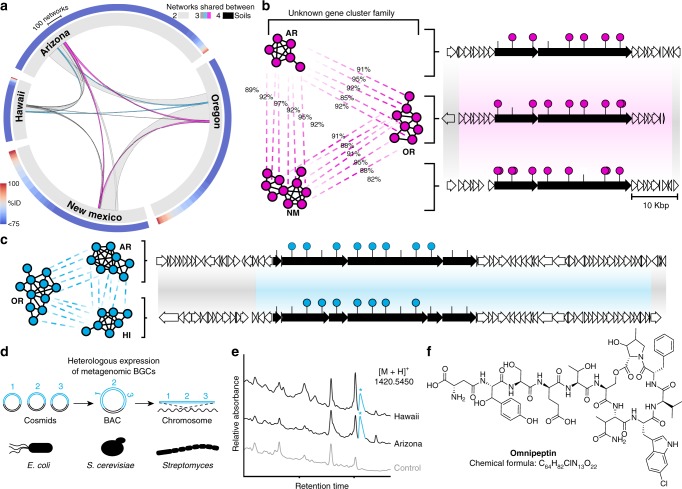
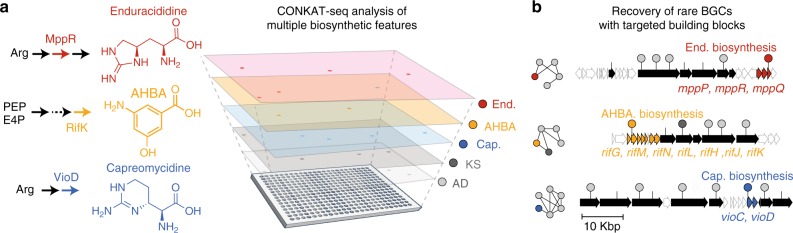
Sequencing of DNA extracted from environmental samples can provide key insights into the biosynthetic potential of uncultured bacteria. However, the high complexity of soil metagenomes, which can contain thousands of bacterial species per gram of soil, imposes significant challenges to explore secondary metabolites potentially produced by rare members of the soil microbiome. Here, we develop a targeted sequencing workflow termed CONKAT-seq (co-occurrence network analysis of targeted sequences) that detects physically clustered biosynthetic domains, a hallmark of bacterial secondary metabolism. Following targeted amplification of conserved biosynthetic domains in a highly partitioned metagenomic library, CONKAT-seq evaluates amplicon co-occurrence patterns across library subpools to identify chromosomally clustered domains. We show that a single soil sample can contain more than a thousand uncharacterized biosynthetic gene clusters, most of which originate from low frequency genomes which are practically inaccessible through untargeted sequencing. CONKAT-seq allows scalable exploration of largely untapped biosynthetic diversity across multiple soils, and can guide the discovery of novel secondary metabolites from rare members of the soil microbiome.
Conflict of interest statement
S.F.B is the founder of LODO Therapeutics. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
