

Hereditary Syndromes with Signs of Premature Aging
- PMID: 31452499
- PMCID: PMC6726857
- DOI: 10.3238/arztebl.2019.0489
Hereditary Syndromes with Signs of Premature Aging
Abstract
Background: Segmental progeroid syndromes (SPS) are rare hereditary diseases in which the affected individuals show signs of premature aging in more than one organ or type of tissue. We review the clinical and genetic features of some of these syndromes and discuss the extent to which their study affords a complementary opportunity to study aging processes in general.
Methods: This review is based on publications retrieved by a selective search in PubMed.
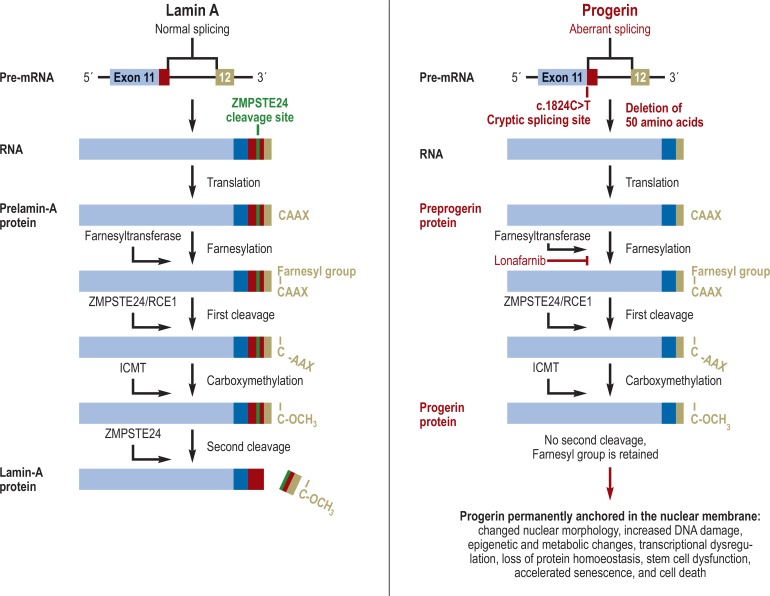
Results: Segmental progeroid syndromes are a clinically and genetically heterogeneous group of hereditary diseases. They can be categorized, for example, by the age of onset of manifestations (congenital vs. infantile vs. juvenile/adult forms). They are diagnosed on clinical grounds supplemented by genetic testing on the basis of next-generation sequencing, which is of central importance in view of the marked heterogeneity and complexity of their overlapping clinical features. The elucidation of the genetic and molecular causes of these diseases can lead to causally directed treatment, as shown by the initial clinical trials in Hutchinson- Gilford progeria syndrome. The molecular features of SPS are identical in many ways to those of "physiological" aging. Thus, studying the molecular mechanisms of SPS may be helpful for the development of molecularly defined treatment approaches for age-associated diseases in general.
Conclusion: Segmental progeroid syndromes are a complex group of diseases with overlapping clinical features. Current research efforts focus on the elucidation of the molecular mechanisms of these diseases, most of which are very rare. This should enable the development of treatments that might be applicable to general processes of aging as well.
Figures
References
-
- Martin GM. Genetic modulation of senescent phenotypes in homo sapiens. Cell. 2005;120:523–532. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources