

Increased expression of heme-binding protein 1 early in Alzheimer's disease is linked to neurotoxicity
- PMID: 31453805
- PMCID: PMC6739868
- DOI: 10.7554/eLife.47498
Increased expression of heme-binding protein 1 early in Alzheimer's disease is linked to neurotoxicity
Abstract
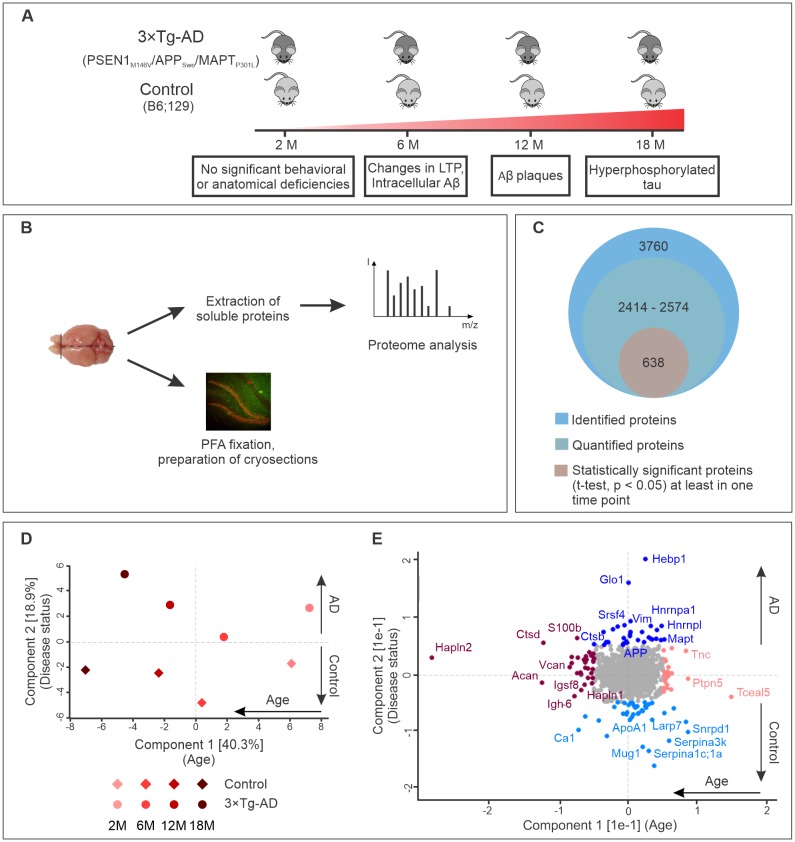
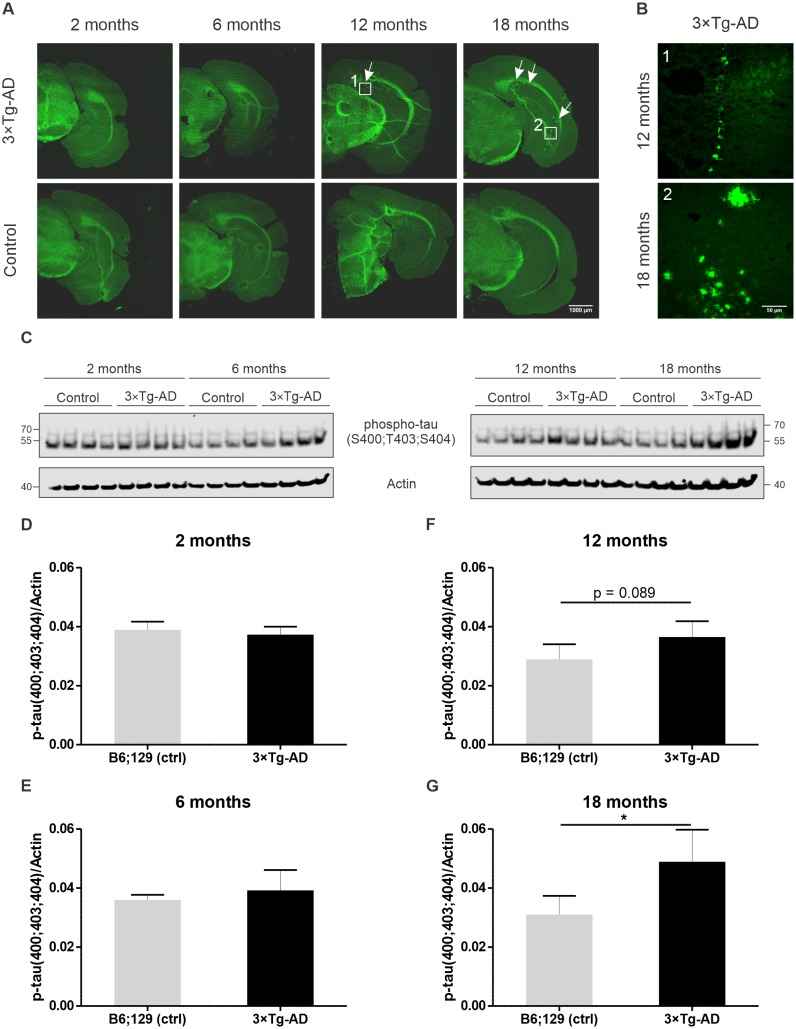
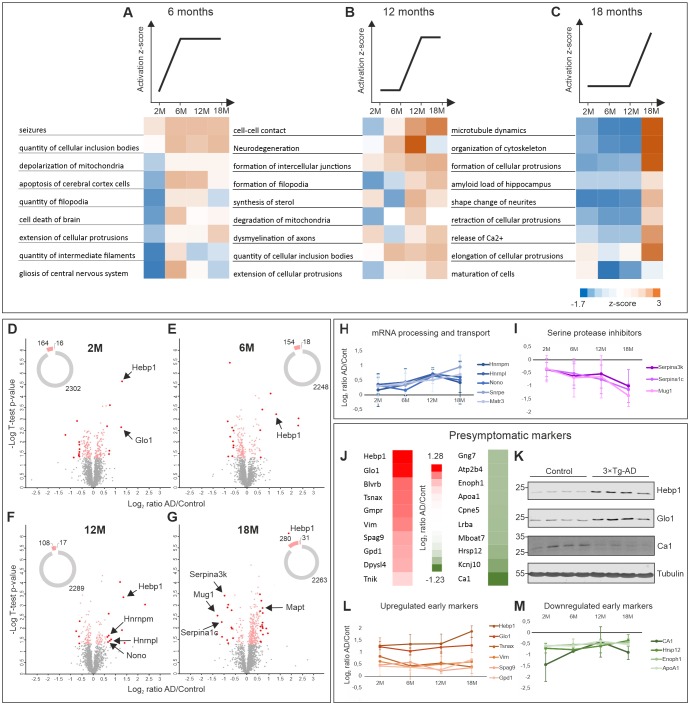
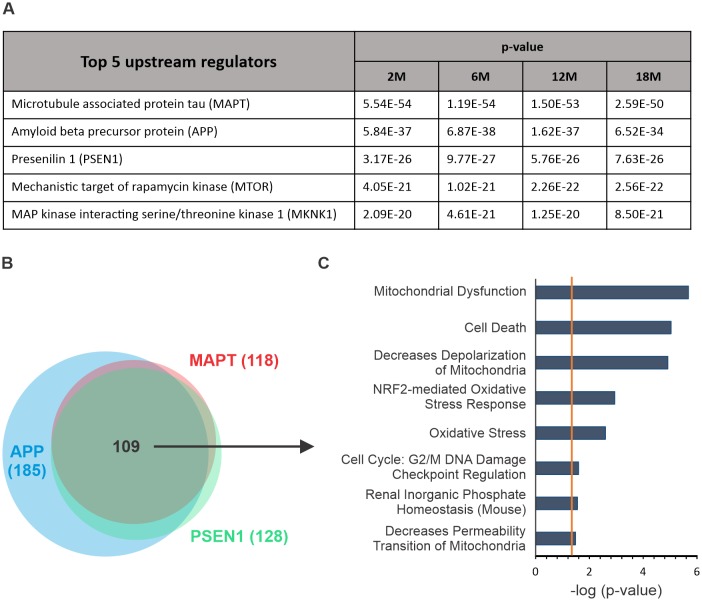
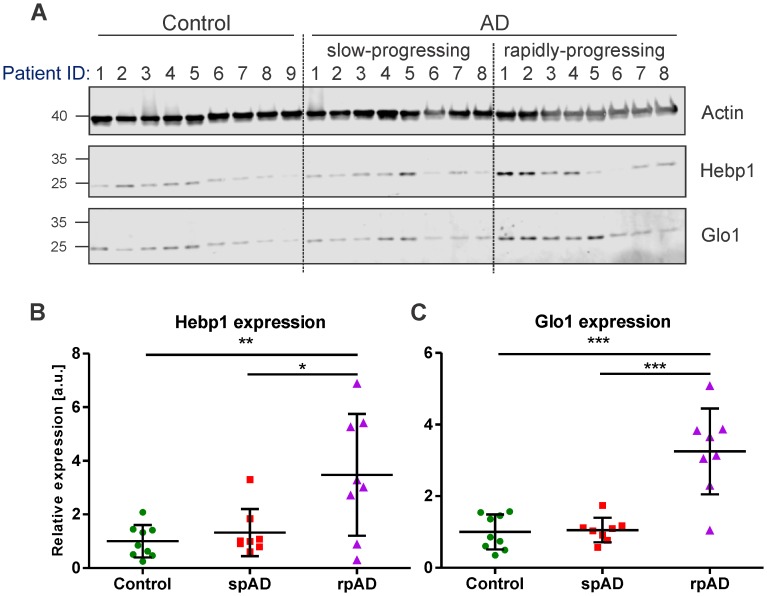
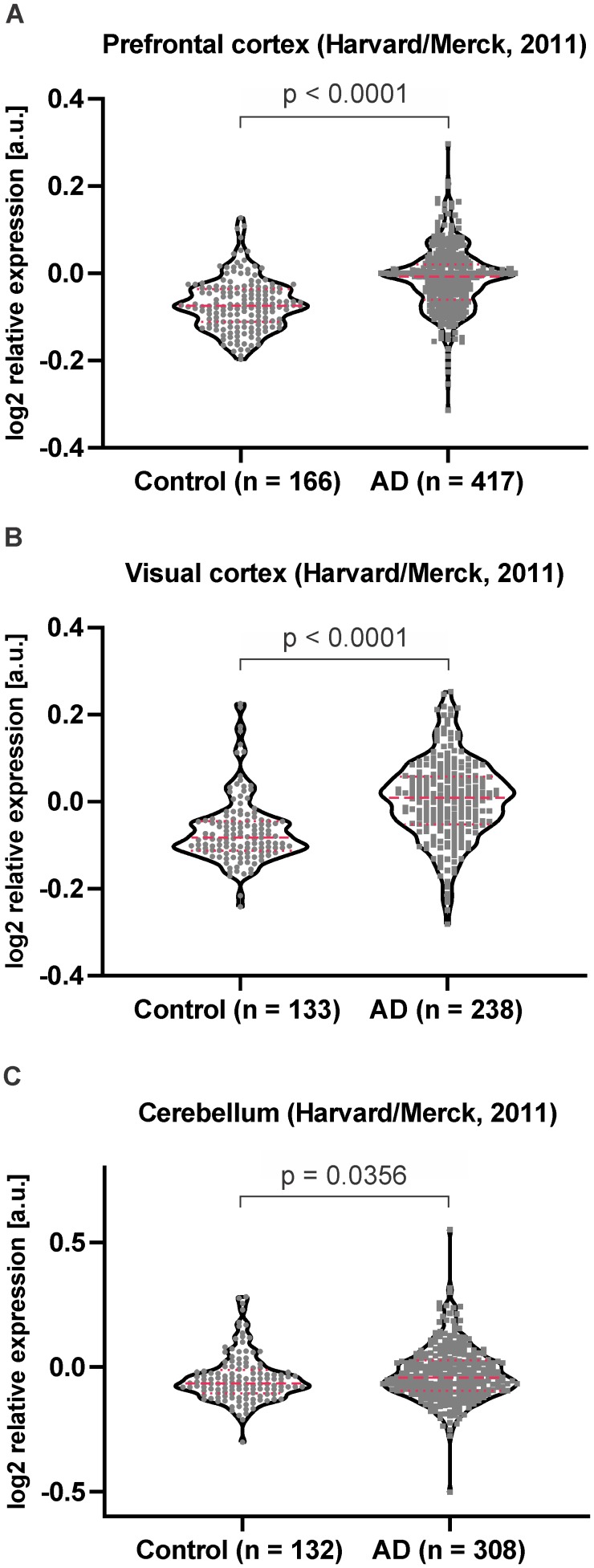
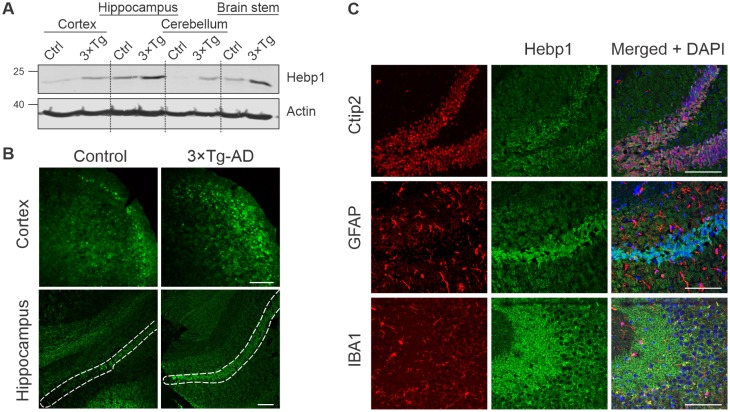
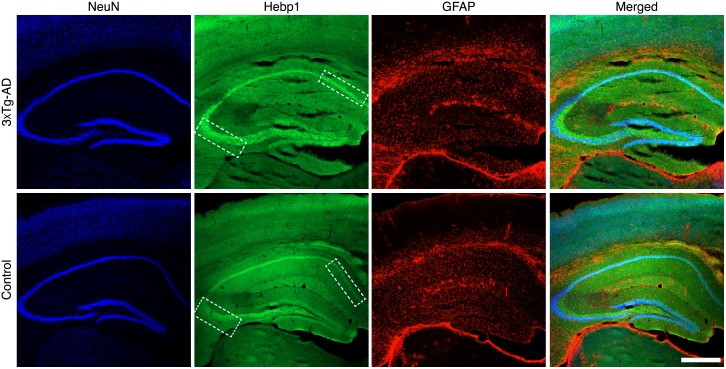
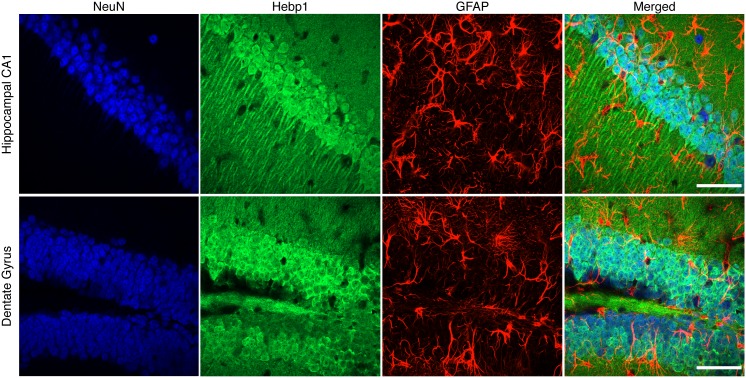
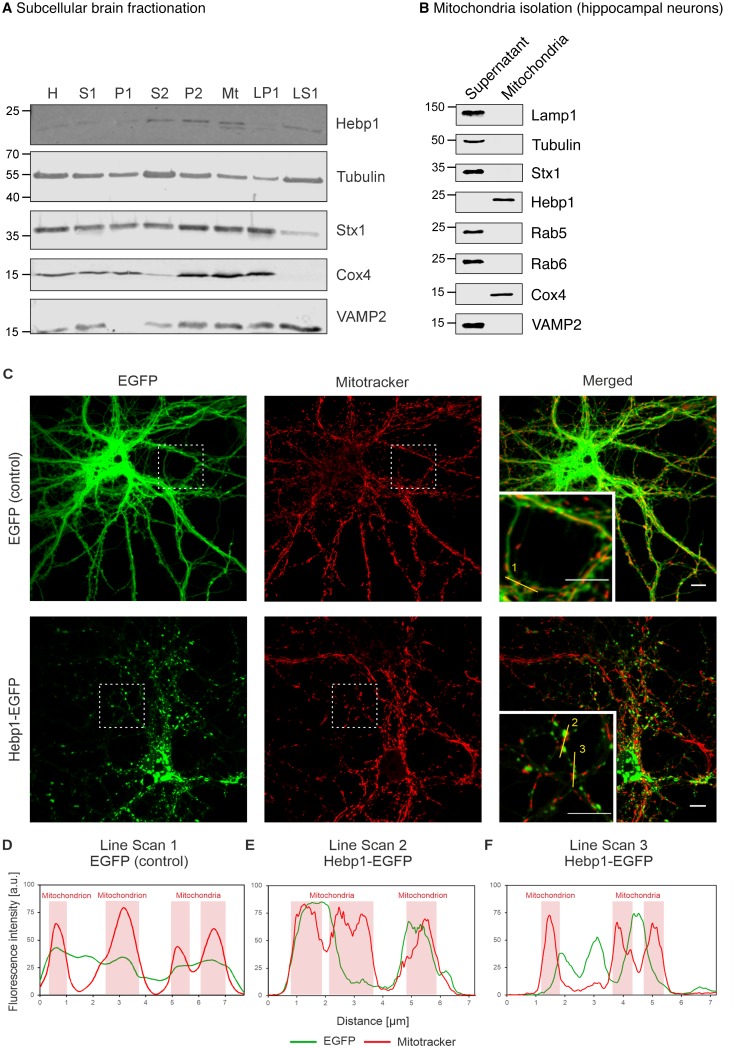
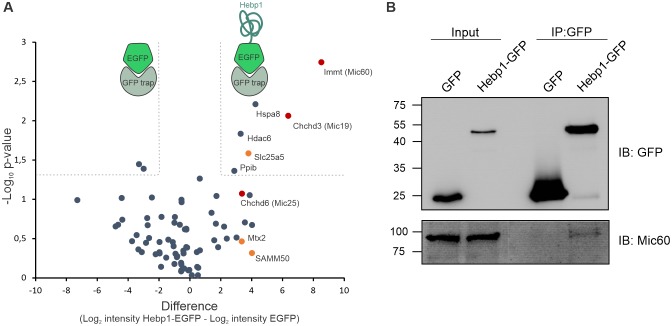
Alzheimer's disease is the most prevalent neurodegenerative disorder leading to progressive cognitive decline. Despite decades of research, understanding AD progression at the molecular level, especially at its early stages, remains elusive. Here, we identified several presymptomatic AD markers by investigating brain proteome changes over the course of neurodegeneration in a transgenic mouse model of AD (3×Tg-AD). We show that one of these markers, heme-binding protein 1 (Hebp1), is elevated in the brains of both 3×Tg-AD mice and patients affected by rapidly-progressing forms of AD. Hebp1, predominantly expressed in neurons, interacts with the mitochondrial contact site complex (MICOS) and exhibits a perimitochondrial localization. Strikingly, wildtype, but not Hebp1-deficient, neurons showed elevated cytotoxicity in response to heme-induced apoptosis. Increased survivability in Hebp1-deficient neurons is conferred by blocking the activation of the mitochondrial-associated caspase signaling pathway. Taken together, our data highlight a role of Hebp1 in progressive neuronal loss during AD progression.
Keywords: Alzheimer's disease; Hebp1; human; mouse; neuron; neuroscience; rat.
© 2019, Yagensky et al.
Conflict of interest statement
OY, MK, SG, TR, SZ, IZ, WH, HU, JC No competing interests declared
Figures
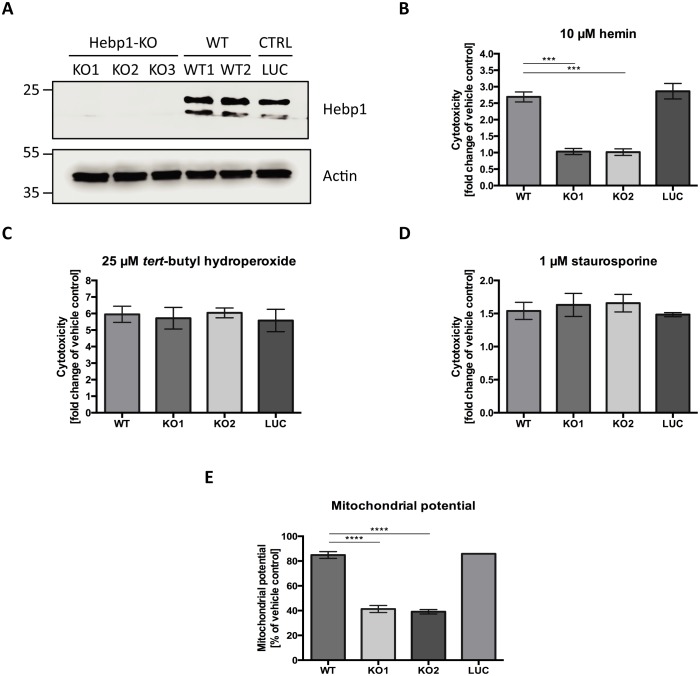
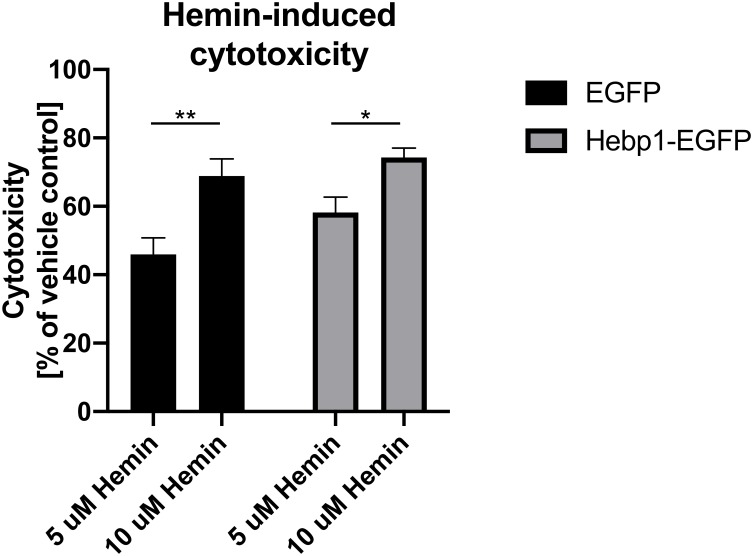
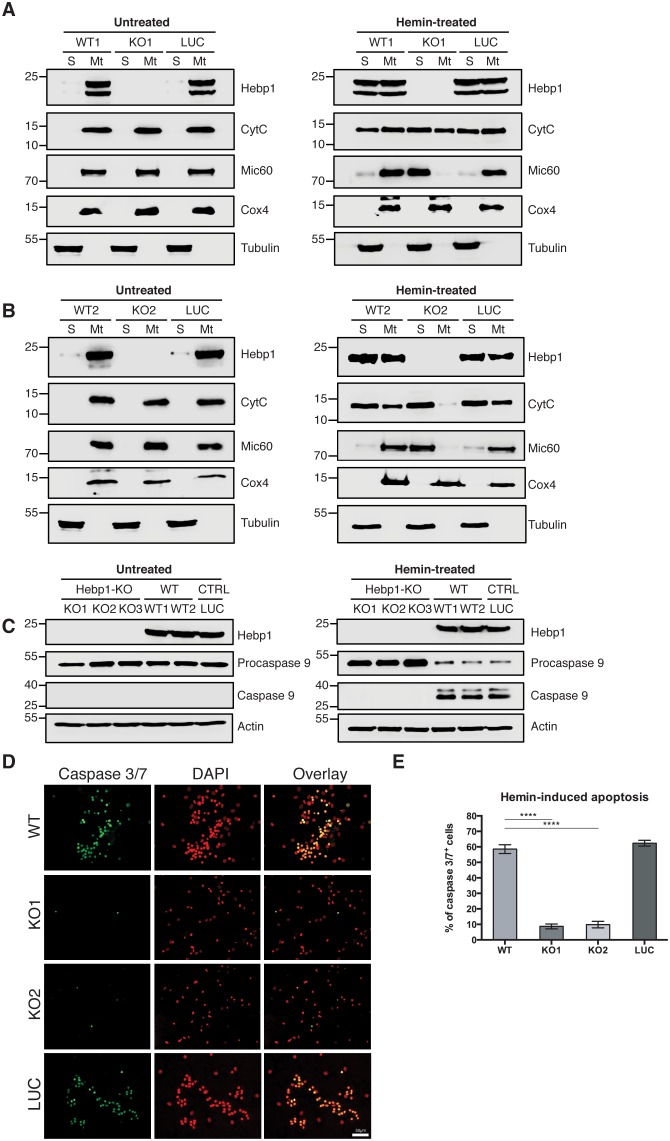
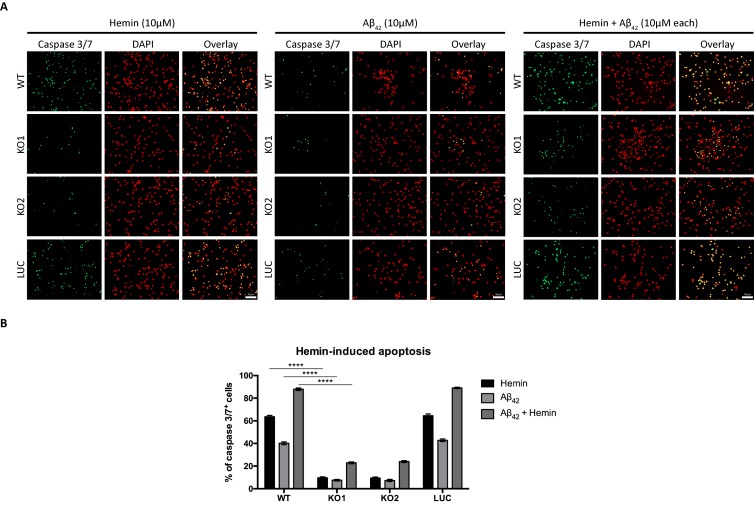
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
