

Clinical, histological, and genetic characterization of PYROXD1-related myopathy
- PMID: 31455395
- PMCID: PMC6710884
- DOI: 10.1186/s40478-019-0781-8
Clinical, histological, and genetic characterization of PYROXD1-related myopathy
Abstract
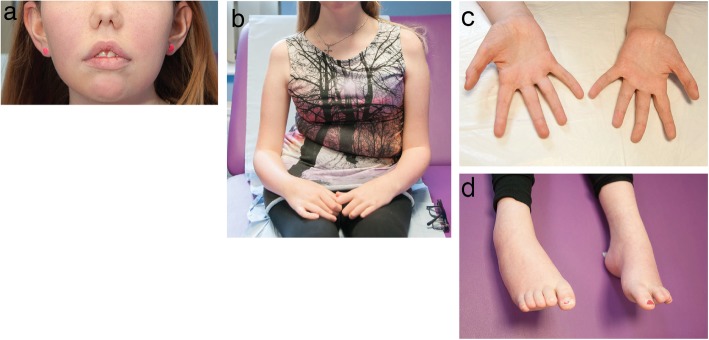
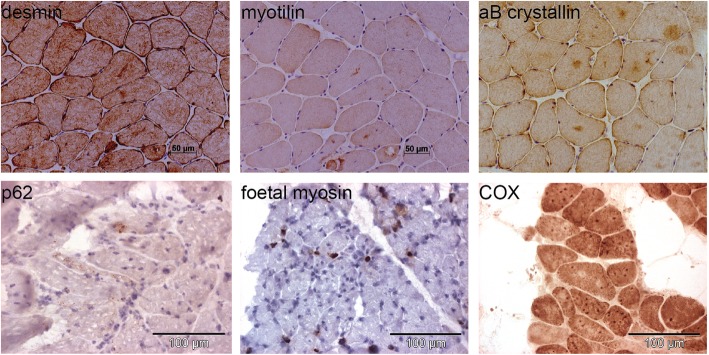
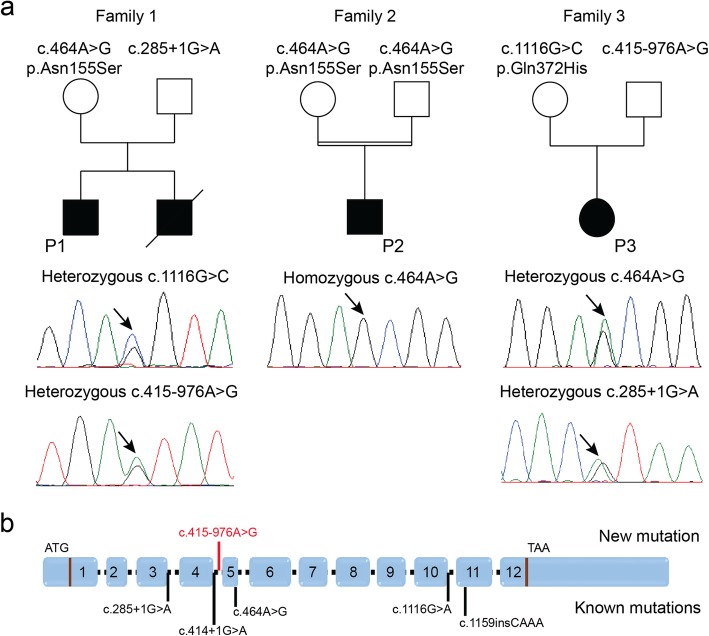
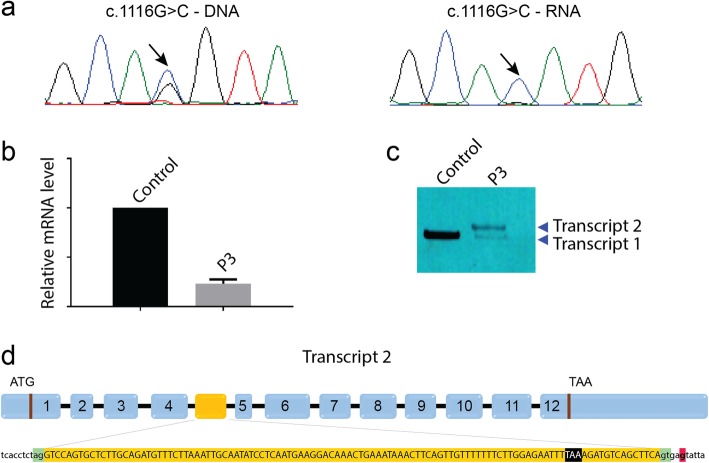
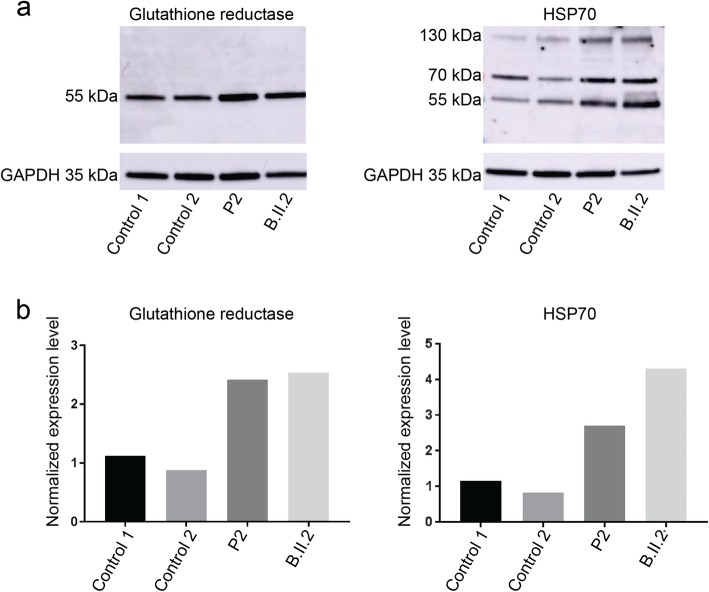
Recessive mutations in PYROXD1, encoding an oxidoreductase, were recently reported in families with congenital myopathy or limb-girdle muscular dystrophy. Here we describe three novel PYROXD1 families at the clinical, histological, and genetic level. Histological analyses on muscle biopsies from all families revealed fiber size variability, endomysial fibrosis, and muscle fibers with multiple internal nuclei and cores. Further characterization of the structural muscle defects uncovered aggregations of myofibrillar proteins, and provided evidence for enhanced oxidative stress. Sequencing identified homozygous or compound heterozygous PYROXD1 mutations including the first deep intronic mutation reinforcing a cryptic donor splice site and resulting in mRNA instability through exonisation of an intronic segment. Overall, this work expands the PYROXD1 mutation spectrum, defines and specifies the histopathological hallmarks of the disorder, and indicates that oxidative stress contributes to the pathomechanism. Comparison of all new and published cases uncovered a genotype/phenotype correlation with a more severe and early-onset phenotypic presentation of patients harboring splice mutations resulting in reduced PYROXD1 protein levels compared with patients carrying missense mutations.
Keywords: Congenital myopathy; LGMD; Myofibrillar inclusions; Oxidoreductase; PYROXD1.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Dubowitz Victor, Sewry Caroline A. Muscle Biopsy. 2007. Normal muscle; pp. 41–74.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
