

Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia
- PMID: 31455887
- PMCID: PMC6791127
- DOI: 10.1038/s41593-019-0463-7
Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia
Abstract
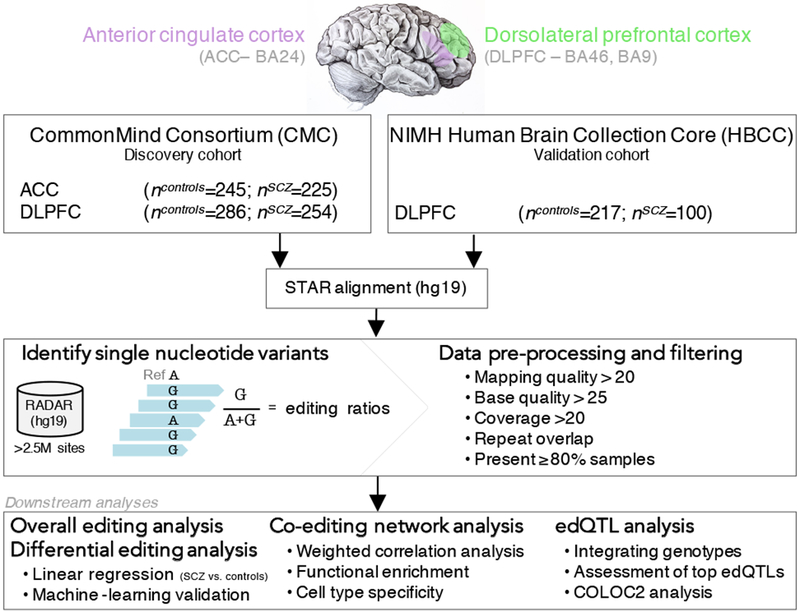
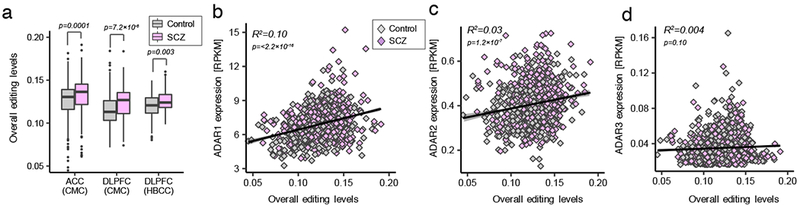
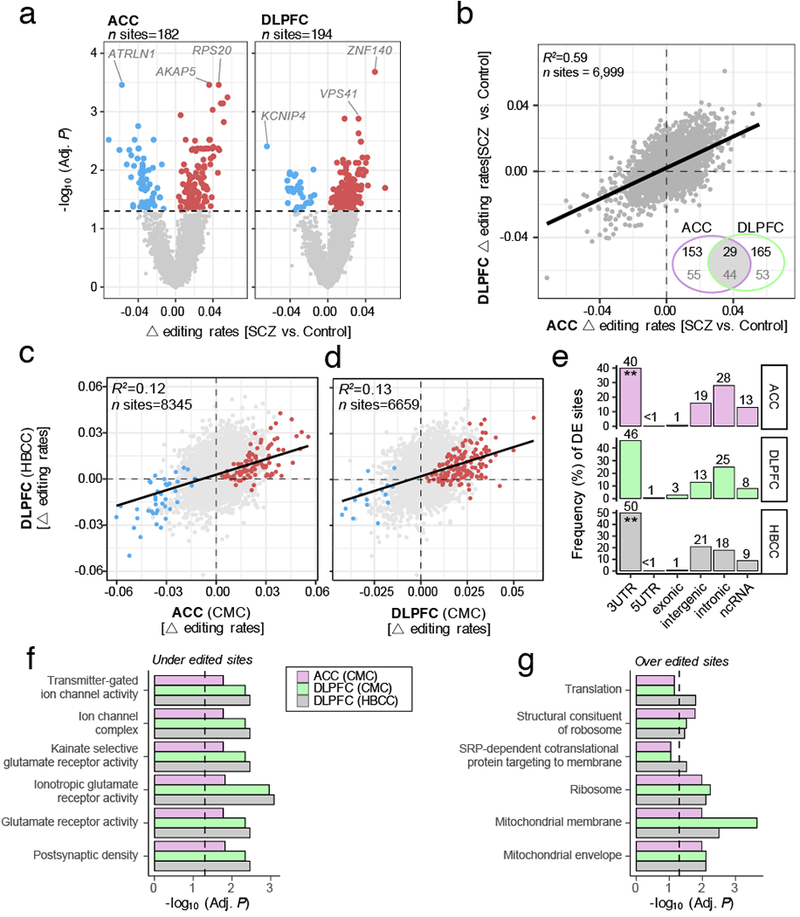
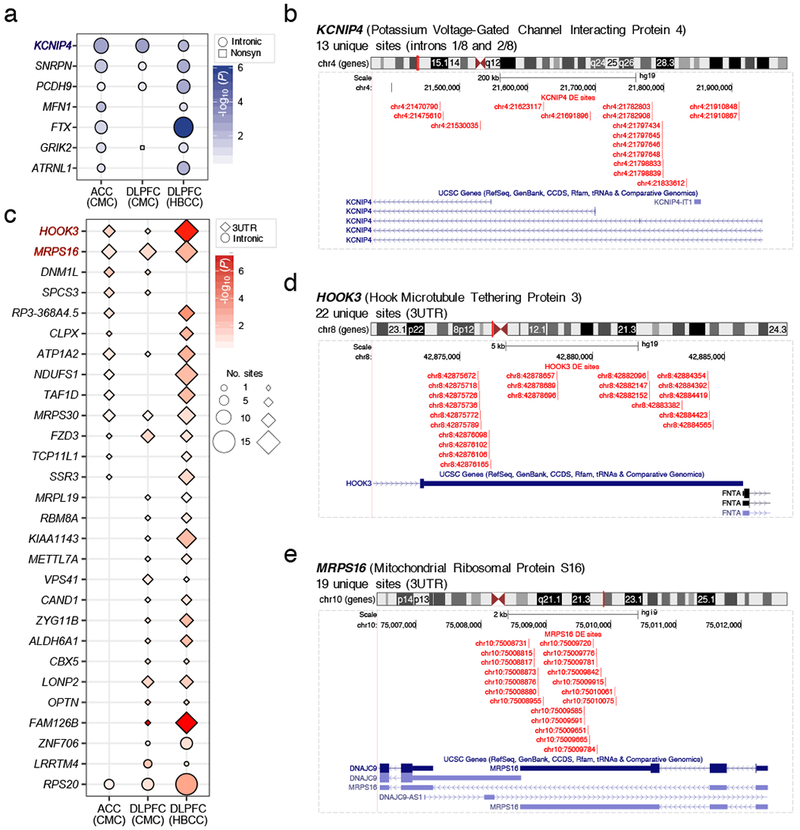
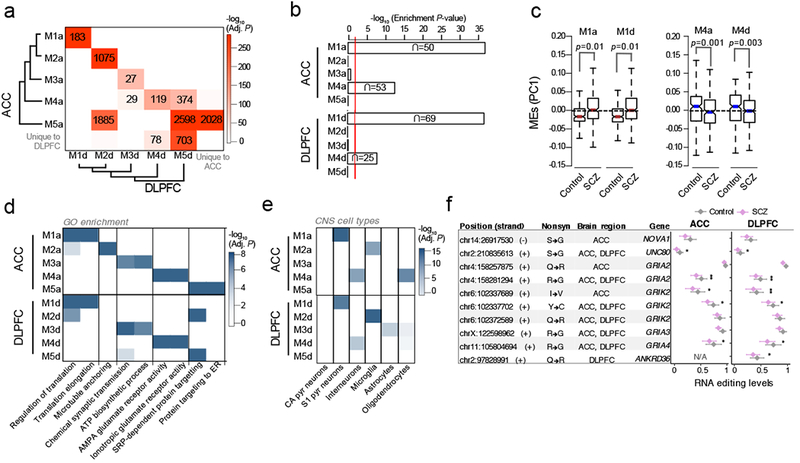
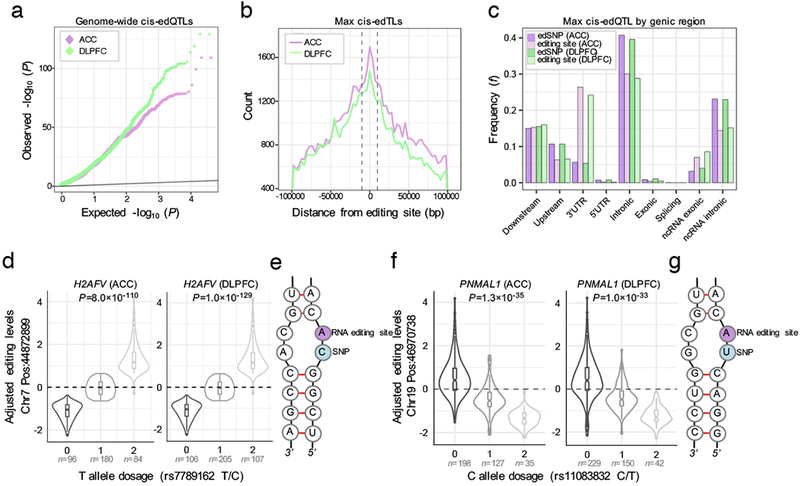
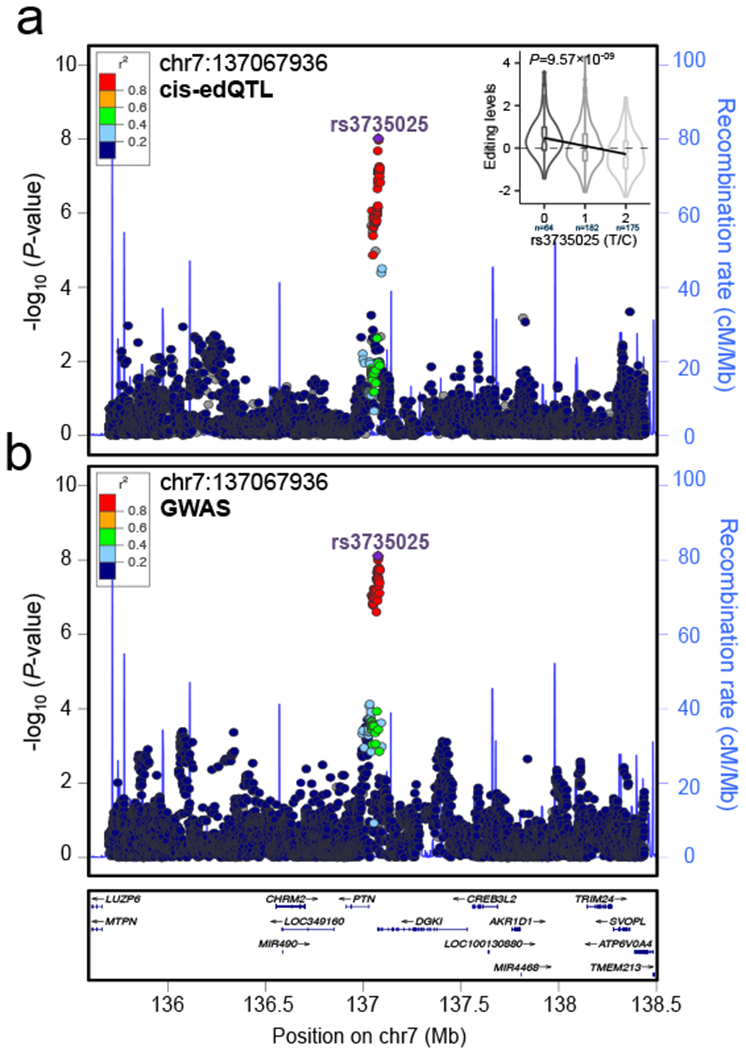
RNA editing critically regulates neurodevelopment and normal neuronal function. The global landscape of RNA editing was surveyed across 364 schizophrenia cases and 383 control postmortem brain samples from the CommonMind Consortium, comprising two regions: dorsolateral prefrontal cortex and anterior cingulate cortex. In schizophrenia, RNA editing sites in genes encoding AMPA-type glutamate receptors and postsynaptic density proteins were less edited, whereas those encoding translation initiation machinery were edited more. These sites replicate between brain regions, map to 3'-untranslated regions and intronic regions, share common sequence motifs and overlap with binding sites for RNA-binding proteins crucial for neurodevelopment. These findings cross-validate in hundreds of non-overlapping dorsolateral prefrontal cortex samples. Furthermore, ~30% of RNA editing sites associate with cis-regulatory variants (editing quantitative trait loci or edQTLs). Fine-mapping edQTLs with schizophrenia risk loci revealed co-localization of eleven edQTLs with six loci. The findings demonstrate widespread altered RNA editing in schizophrenia and its genetic regulation, and suggest a causal and mechanistic role of RNA editing in schizophrenia neuropathology.
Conflict of interest statement
COMPETING INTERESTS
None to declare.
Figures
References
REFERENCES (materials and methods)
-
- Buuren SV & Groothuis-Oudshoorn K, mice: Multivariate imputation by chained equations in R. Journal of statistical software 45, 1–68 (2010).
Publication types
MeSH terms
Grants and funding
- U01 MH116442/MH/NIMH NIH HHS/United States
- R01 MH109897/MH/NIMH NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- R01 MH074313/MH/NIMH NIH HHS/United States
- U01 MH103392/MH/NIMH NIH HHS/United States
- U01 MH111661/MH/NIMH NIH HHS/United States
- P50 MH084053/MH/NIMH NIH HHS/United States
- HHSN271201300031C/MH/NIMH NIH HHS/United States
- R01 MH110921/MH/NIMH NIH HHS/United States
- P50 MH066392/MH/NIMH NIH HHS/United States
- R01 MH080405/MH/NIMH NIH HHS/United States
- R01 MH085542/MH/NIMH NIH HHS/United States
- R01 MH075916/MH/NIMH NIH HHS/United States
- T32 AG049688/AG/NIA NIH HHS/United States
- R01 MH057881/MH/NIMH NIH HHS/United States
- S10 OD018522/OD/NIH HHS/United States
- R01 MH109677/MH/NIMH NIH HHS/United States
- R01 MH097276/MH/NIMH NIH HHS/United States
- P01 AG002219/AG/NIA NIH HHS/United States
- R01 MH093725/MH/NIMH NIH HHS/United States
- P50 AG005138/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
