

The Pyrin Inflammasome in Health and Disease
- PMID: 31456795
- PMCID: PMC6698799
- DOI: 10.3389/fimmu.2019.01745
The Pyrin Inflammasome in Health and Disease
Abstract
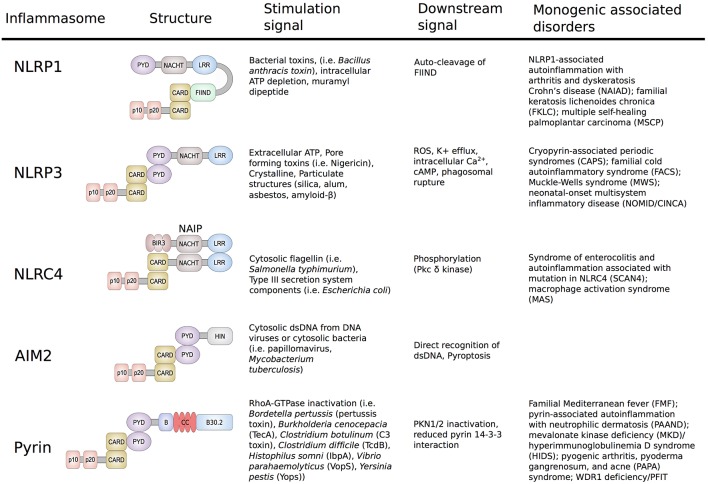
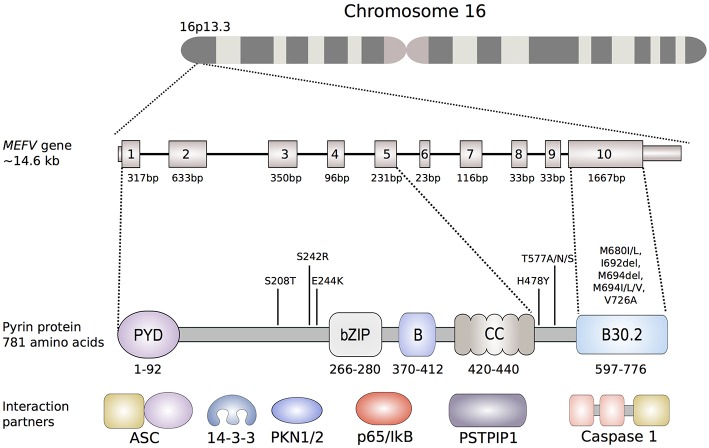
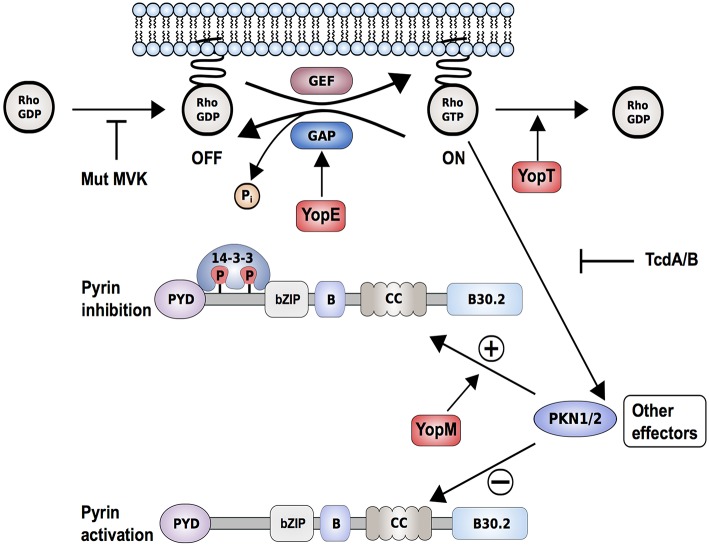
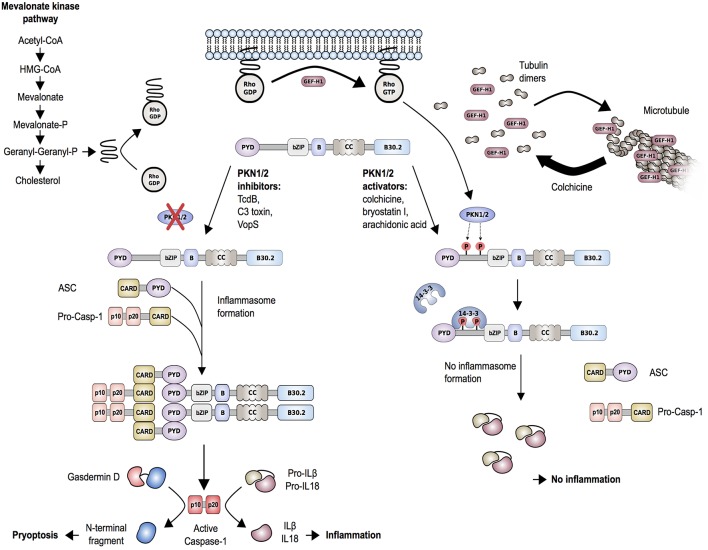
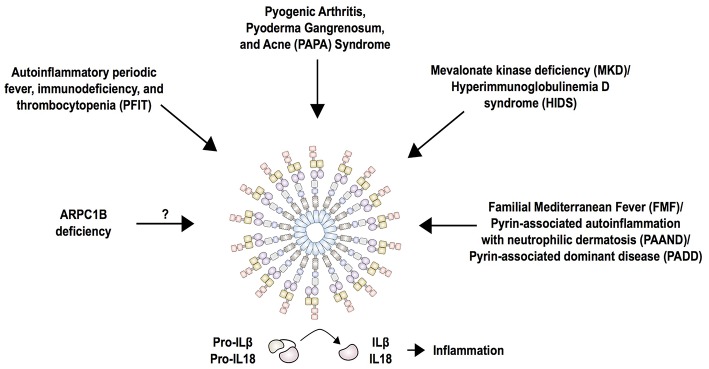
The pyrin inflammasome has evolved as an innate immune sensor to detect bacterial toxin-induced Rho guanosine triphosphatase (Rho GTPase)-inactivation, a process that is similar to the "guard" mechanism in plants. Rho GTPases act as molecular switches to regulate a variety of signal transduction pathways including cytoskeletal organization. Pathogens can modulate Rho GTPase activity to suppress host immune responses such as phagocytosis. Pyrin is encoded by MEFV, the gene that is mutated in patients with familial Mediterranean fever (FMF). FMF is the prototypic autoinflammatory disease characterized by recurring short episodes of systemic inflammation and is a common disorder in many populations in the Mediterranean basin. Pyrin specifically senses modifications in the activity of the small GTPase RhoA, which binds to many effector proteins including the serine/threonine-protein kinases PKN1 and PKN2 and actin-binding proteins. RhoA activation leads to PKN-mediated phosphorylation-dependent pyrin inhibition. Conversely, pathogen virulence factors downregulate RhoA activity in a variety of ways, and these changes are detected by the pyrin inflammasome irrespective of the type of modifications. MEFV pathogenic variants favor the active state of pyrin and elicit proinflammatory cytokine release and pyroptosis. They can be inherited either as a dominant or recessive trait depending on the variant's location and effect on the protein function. Mutations in the C-terminal B30.2 domain are usually considered recessive, although heterozygotes may manifest a biochemical or even a clinical phenotype. These variants are hypomorphic in regard to their effect on intramolecular interactions, but ultimately accentuate pyrin activity. Heterozygous mutations in other domains of pyrin affect residues critical for inhibition or protein oligomerization, and lead to constitutively active inflammasome. In healthy carriers of FMF mutations who have the subclinical inflammatory phenotype, the increased activity of pyrin might have been protective against endemic infections over human history. This finding is supported by the observation of high carrier frequencies of FMF-mutations in multiple populations. The pyrin inflammasome also plays a role in mediating inflammation in other autoinflammatory diseases linked to dysregulation in the actin polymerization pathway. Therefore, the assembly of the pyrin inflammasome is initiated in response to fluctuations in cytoplasmic homeostasis and perturbations in cytoskeletal dynamics.
Keywords: RhoA GTPases; Yersinia toxins; autoinflammatory diseases; familial Mediterranean fever; pyrin inflammasome; serine-threonine kinase.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources