

Glucocerebrosidase and its relevance to Parkinson disease
- PMID: 31464647
- PMCID: PMC6716912
- DOI: 10.1186/s13024-019-0336-2
Glucocerebrosidase and its relevance to Parkinson disease
Abstract
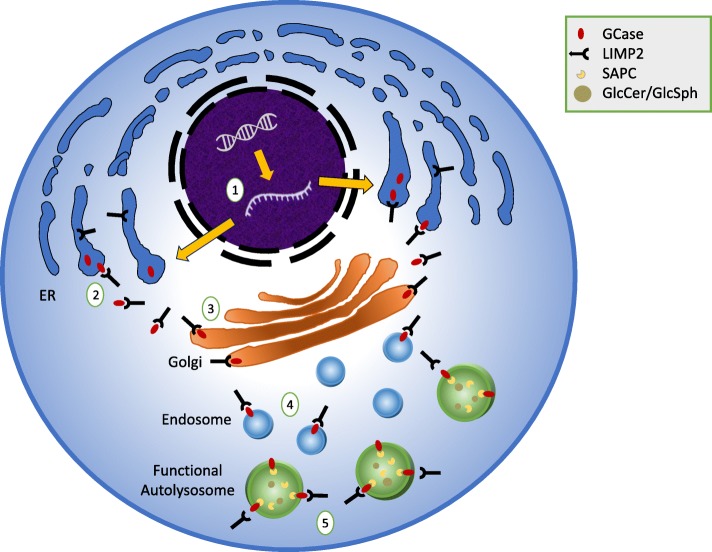
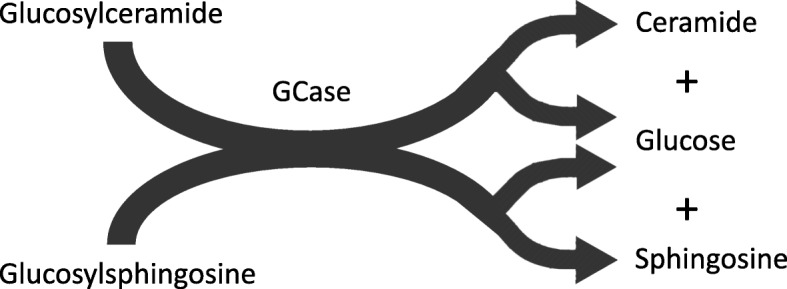
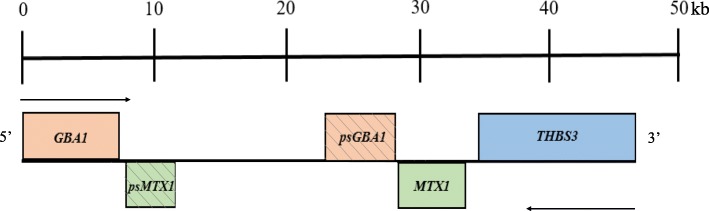
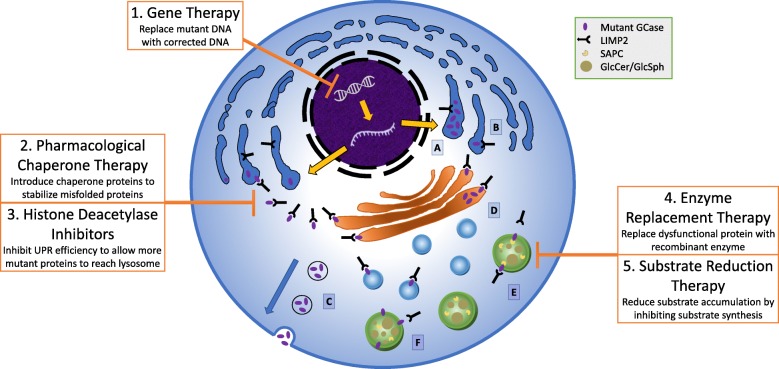
Mutations in GBA1, the gene encoding the lysosomal enzyme glucocerebrosidase, are among the most common known genetic risk factors for the development of Parkinson disease and related synucleinopathies. A great deal is known about GBA1, as mutations in GBA1 are causal for the rare autosomal storage disorder Gaucher disease. Over the past decades, significant progress has been made in understanding the genetics and cell biology of glucocerebrosidase. A least 495 different mutations, found throughout the 11 exons of the gene are reported, including both common and rare variants. Mutations in GBA1 may lead to degradation of the protein, disruptions in lysosomal targeting and diminished performance of the enzyme in the lysosome.Gaucher disease is phenotypically diverse and has both neuronopathic and non-neuronopathic forms. Both patients with Gaucher disease and heterozygous carriers are at increased risk of developing Parkinson disease and Dementia with Lewy Bodies, although our understanding of the mechanism for this association remains incomplete. There appears to be an inverse relationship between glucocerebrosidase and α-synuclein levels, and even patients with sporadic Parkinson disease have decreased glucocerebrosidase. Glucocerebrosidase may interact with α-synuclein to maintain basic cellular functions, or impaired glucocerebrosidase could contribute to Parkinson pathogenesis by disrupting lysosomal homeostasis, enhancing endoplasmic reticulum stress or contributing to mitochondrial impairment. However, the majority of patients with GBA1 mutations never develop parkinsonism, so clearly other risk factors play a role. Treatments for Gaucher disease have been developed that increase visceral glucocerebrosidase levels and decrease lipid storage, although they have yet to properly address the neurological defects associated with impaired glucocerebrosidase. Mouse and induced pluripotent stem cell derived models have improved our understanding of glucocerebrosidase function and the consequences of its deficiency. These models have been used to test novel therapies including chaperone proteins, histone deacetylase inhibitors, and gene therapy approaches that enhance glucocerebrosidase levels and could prove efficacious in the treatment of forms of parkinsonism. Consequently, this rare monogenic disorder, Gaucher disease, provides unique insights directly applicable to our understanding and treatment of Parkinson disease, a common and complex neurodegenerative disorder.
Keywords: GBA1; Gaucher disease; Glucocerebrosidase; Lysosome; Parkinson disease; α-Synuclein.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Gaucher P. De l’epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucmie. 1882.
-
- Oberling C. Rev franc de Ped III. 1927.
-
- Brady RO, Kanfer J, Shapiro D. The metabolism of Glucocerebrosides. I. Purification and properties of a Glucocerebroside-cleaving enzyme from spleen tissue. J Biol Chem. 1965;240:39–43. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
