

Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease
- PMID: 31467276
- PMCID: PMC6715724
- DOI: 10.1038/s41467-019-11813-6
Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease
Abstract
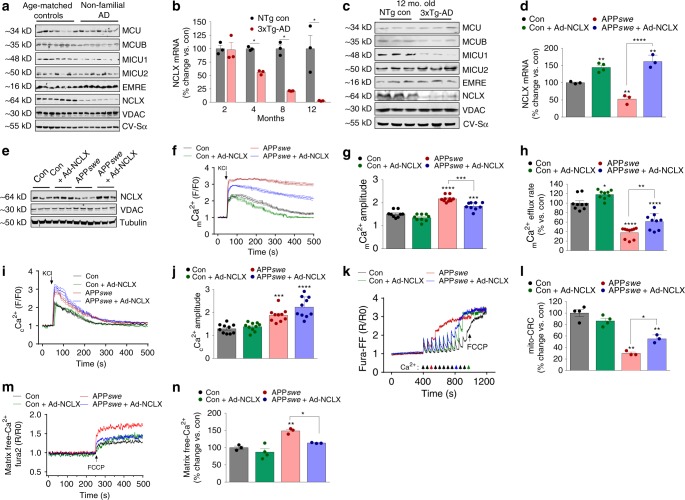
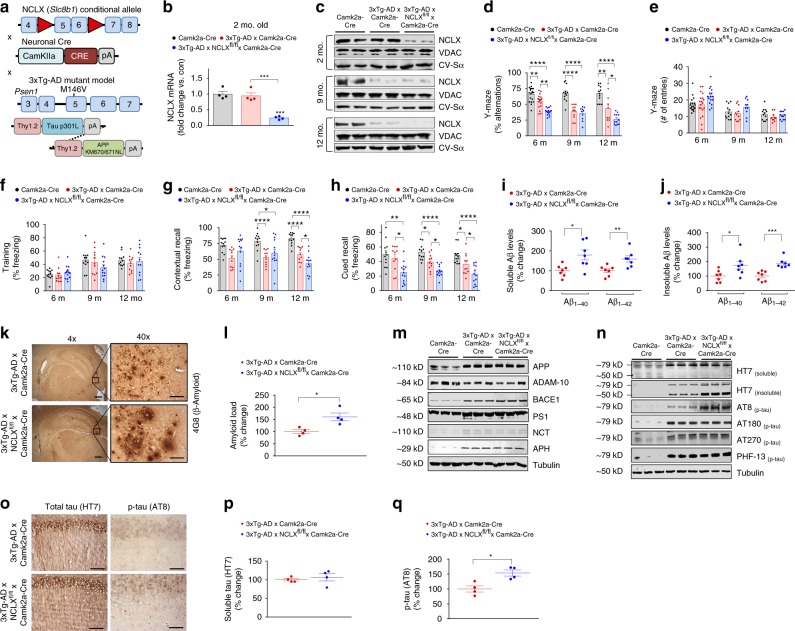
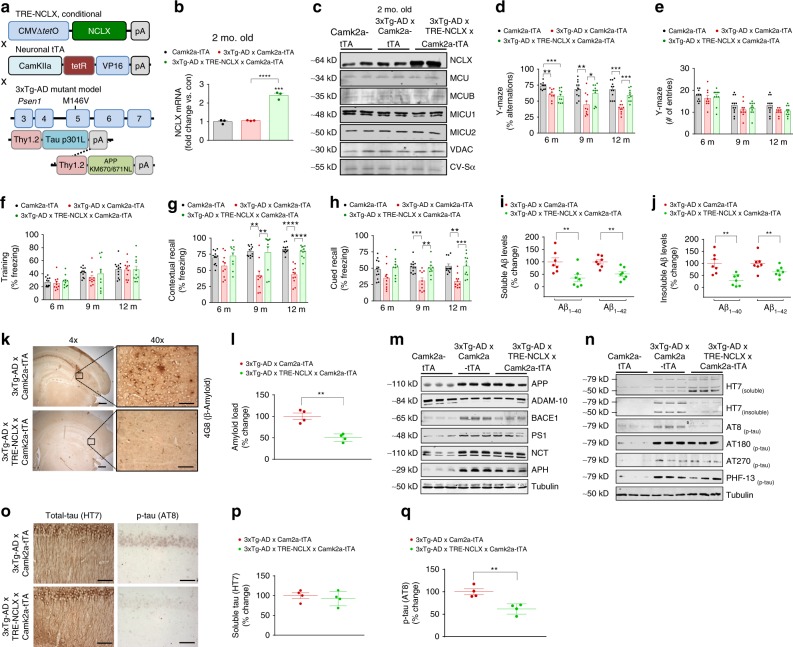
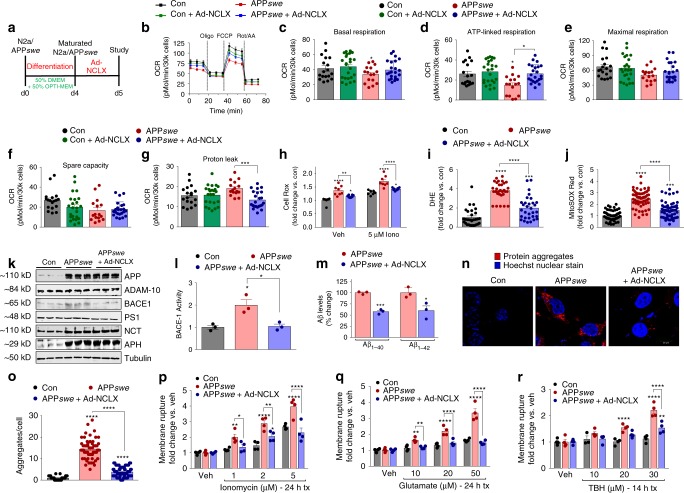
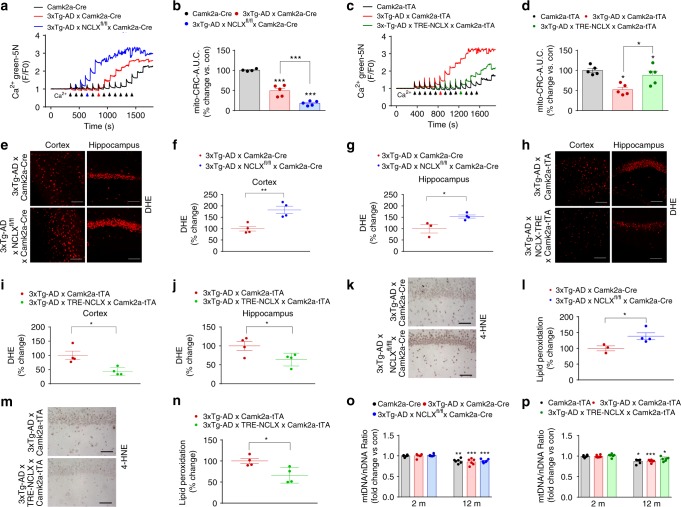
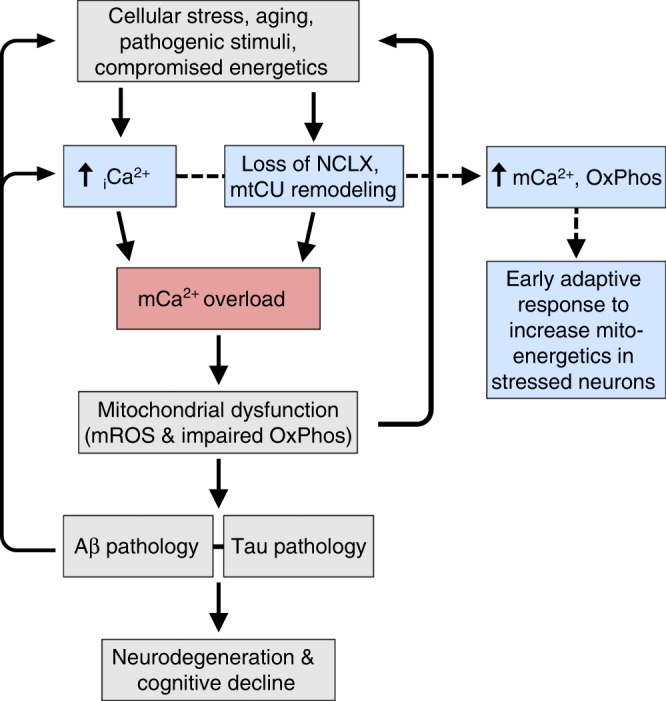
Impairments in neuronal intracellular calcium (iCa2+) handling may contribute to Alzheimer's disease (AD) development. Metabolic dysfunction and progressive neuronal loss are associated with AD progression, and mitochondrial calcium (mCa2+) signaling is a key regulator of both of these processes. Here, we report remodeling of the mCa2+ exchange machinery in the prefrontal cortex of individuals with AD. In the 3xTg-AD mouse model impaired mCa2+ efflux capacity precedes neuropathology. Neuronal deletion of the mitochondrial Na+/Ca2+ exchanger (NCLX, Slc8b1 gene) accelerated memory decline and increased amyloidosis and tau pathology. Further, genetic rescue of neuronal NCLX in 3xTg-AD mice is sufficient to impede AD-associated pathology and memory loss. We show that mCa2+ overload contributes to AD progression by promoting superoxide generation, metabolic dysfunction and neuronal cell death. These results provide a link between the calcium dysregulation and metabolic dysfunction hypotheses of AD and suggest mCa2+ exchange as potential therapeutic target in AD.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Cummings J, et al. Anti-Tau trials for Alzheimer's disease: a report from the EU/US/CTAD task force. J. Prev. Alzheimer's Dis. 2019;6:157–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
