

CSL controls telomere maintenance and genome stability in human dermal fibroblasts
- PMID: 31467287
- PMCID: PMC6715699
- DOI: 10.1038/s41467-019-11785-7
CSL controls telomere maintenance and genome stability in human dermal fibroblasts
Abstract
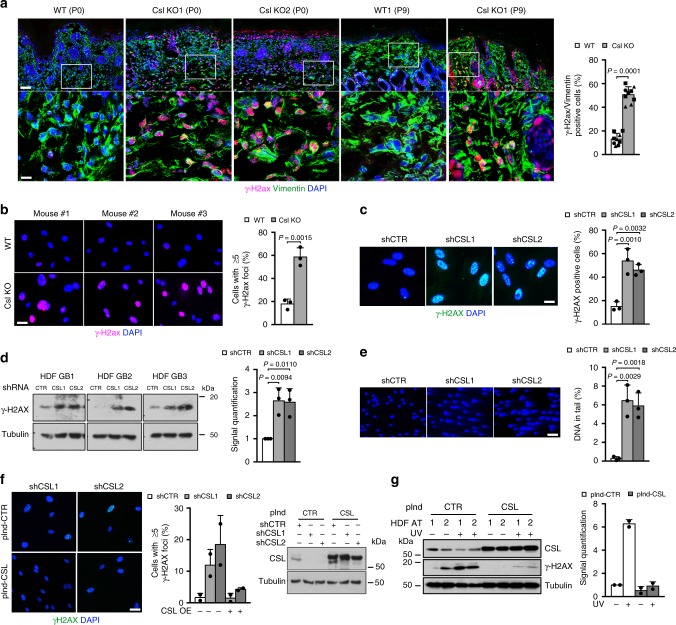
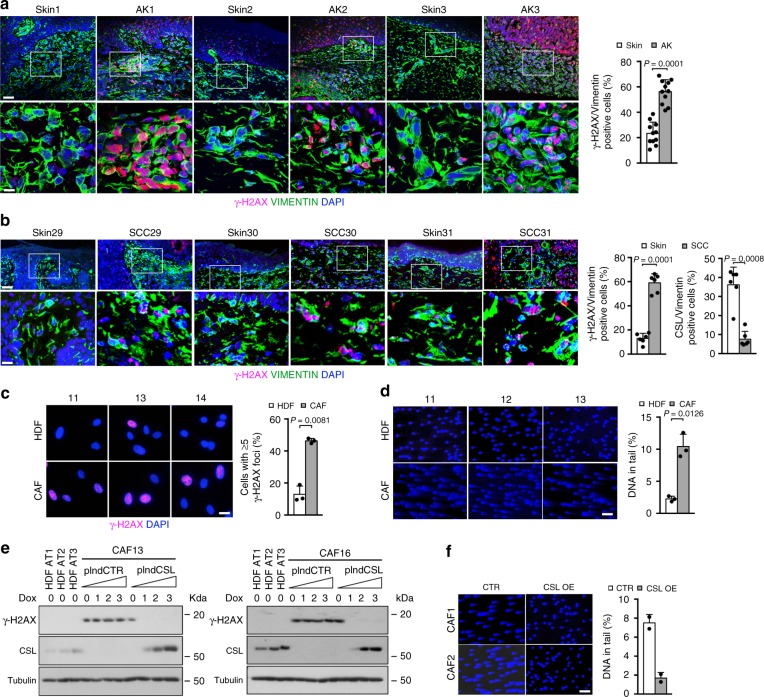
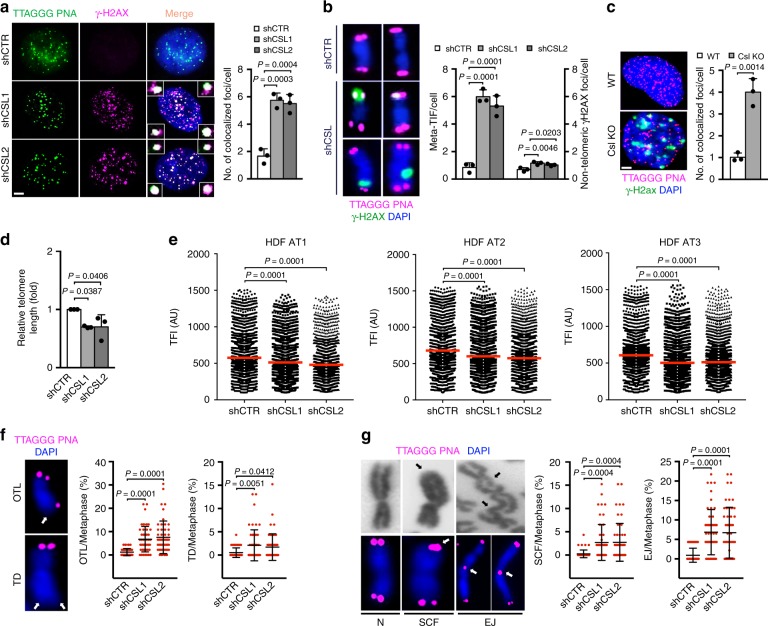
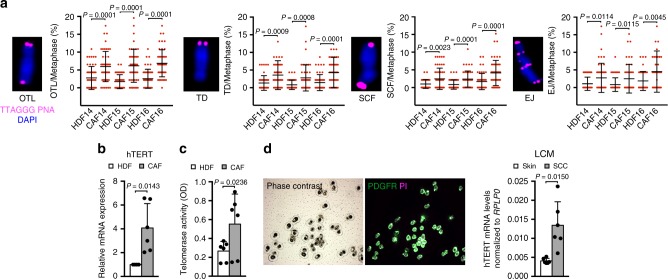
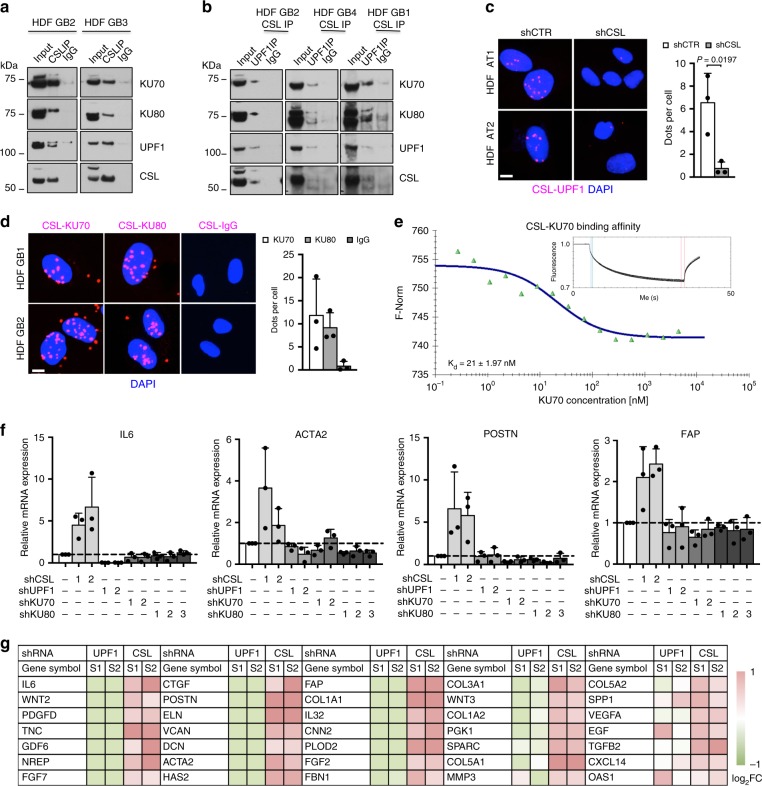
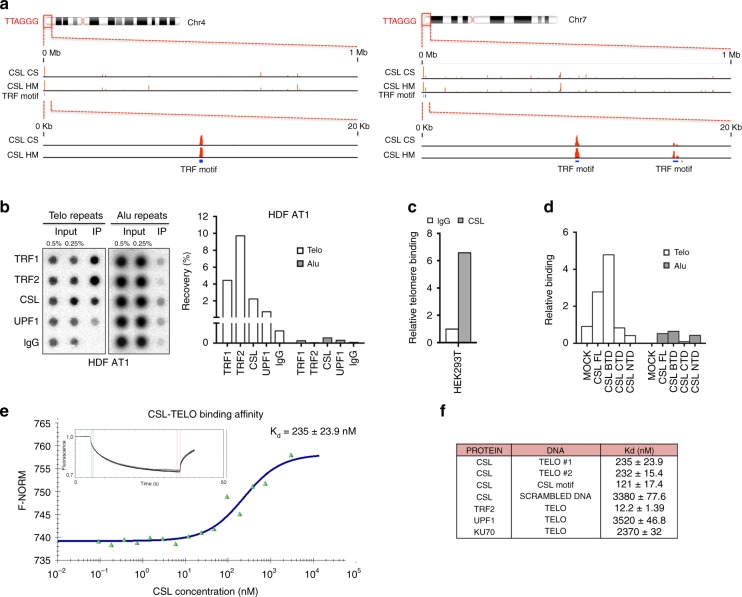
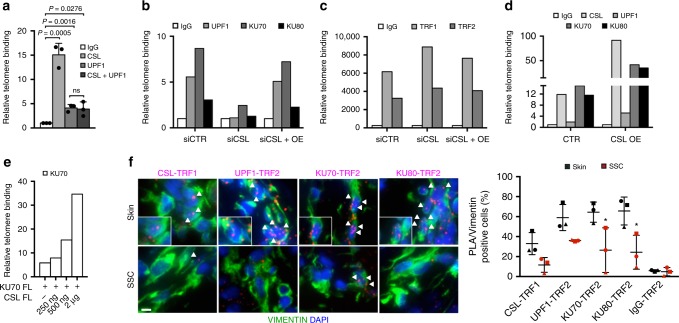
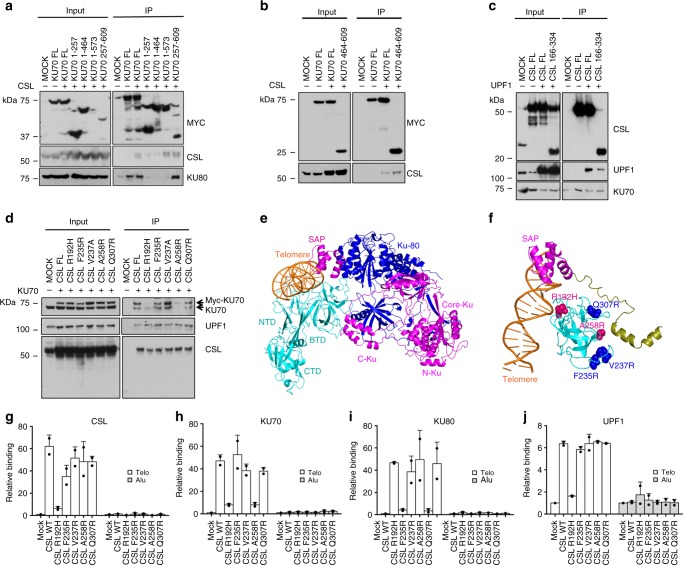
Genomic instability is a hallmark of cancer. Whether it also occurs in Cancer Associated Fibroblasts (CAFs) remains to be carefully investigated. Loss of CSL/RBP-Jκ, the effector of canonical NOTCH signaling with intrinsic transcription repressive function, causes conversion of dermal fibroblasts into CAFs. Here, we find that CSL down-modulation triggers DNA damage, telomere loss and chromosome end fusions that also occur in skin Squamous Cell Carcinoma (SCC)-associated CAFs, in which CSL is decreased. Separately from its role in transcription, we show that CSL is part of a multiprotein telomere protective complex, binding directly and with high affinity to telomeric DNA as well as to UPF1 and Ku70/Ku80 proteins and being required for their telomere association. Taken together, the findings point to a central role of CSL in telomere homeostasis with important implications for genomic instability of cancer stromal cells and beyond.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
