

Microglia Mediate HIV-1 gp120-Induced Synaptic Degeneration in Spinal Pain Neural Circuits
- PMID: 31471472
- PMCID: PMC6794928
- DOI: 10.1523/JNEUROSCI.2851-18.2019
Microglia Mediate HIV-1 gp120-Induced Synaptic Degeneration in Spinal Pain Neural Circuits
Abstract
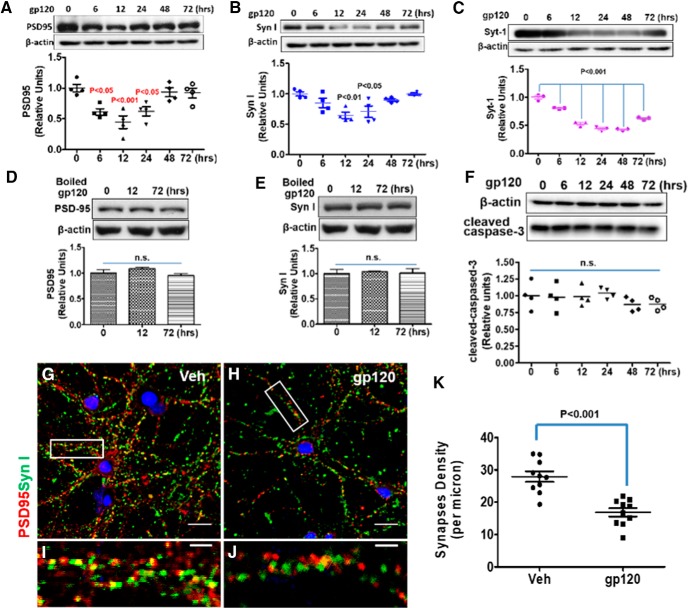
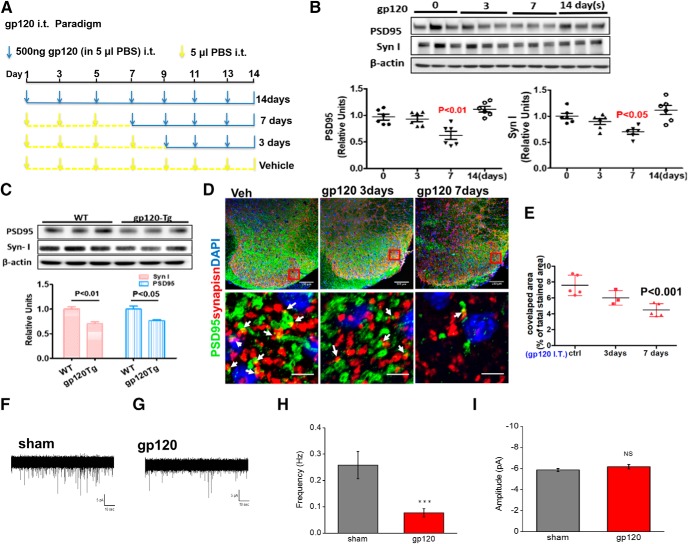
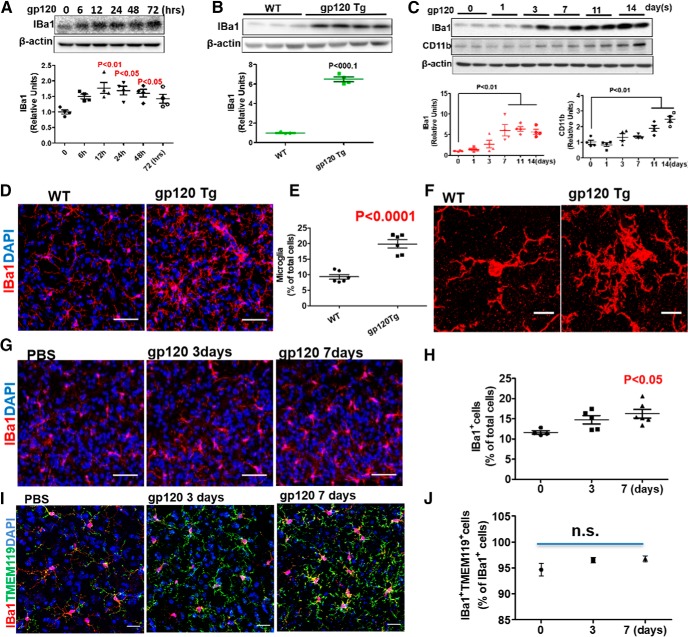
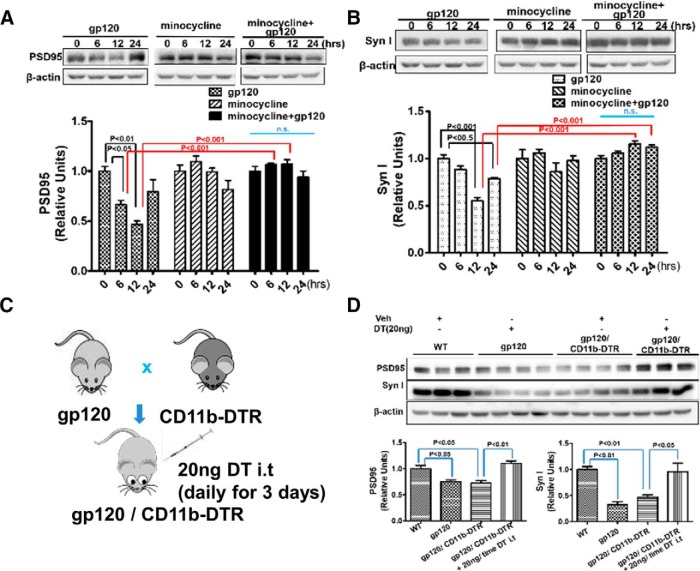
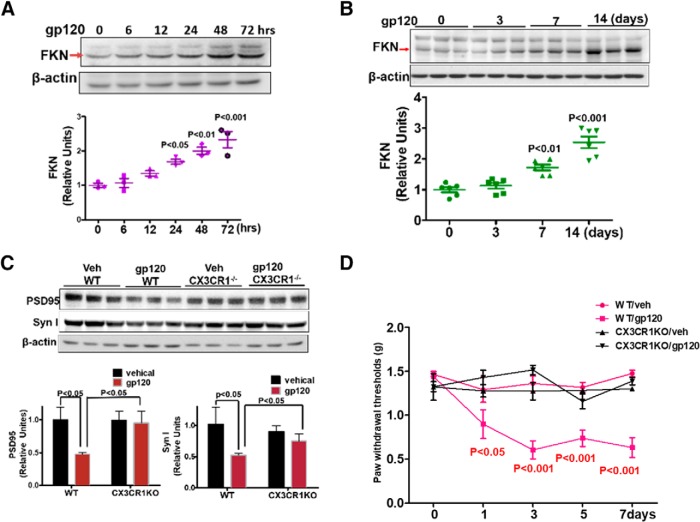
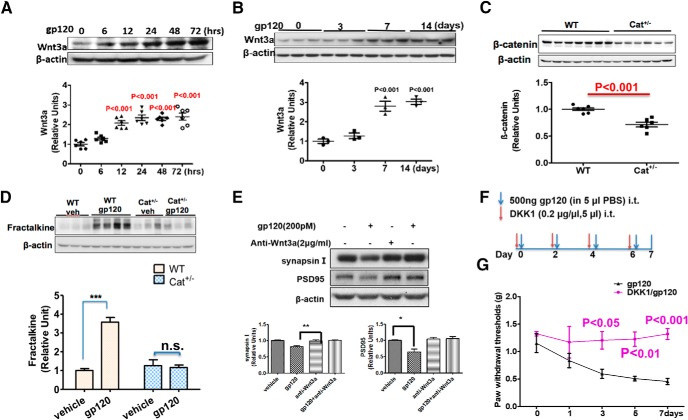
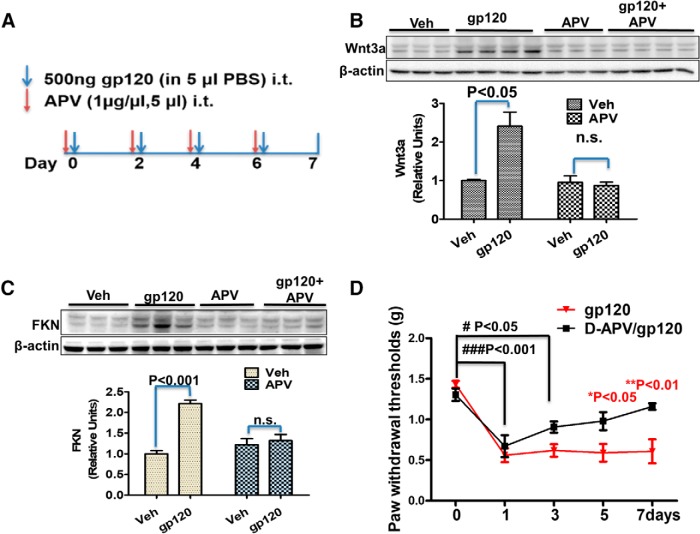
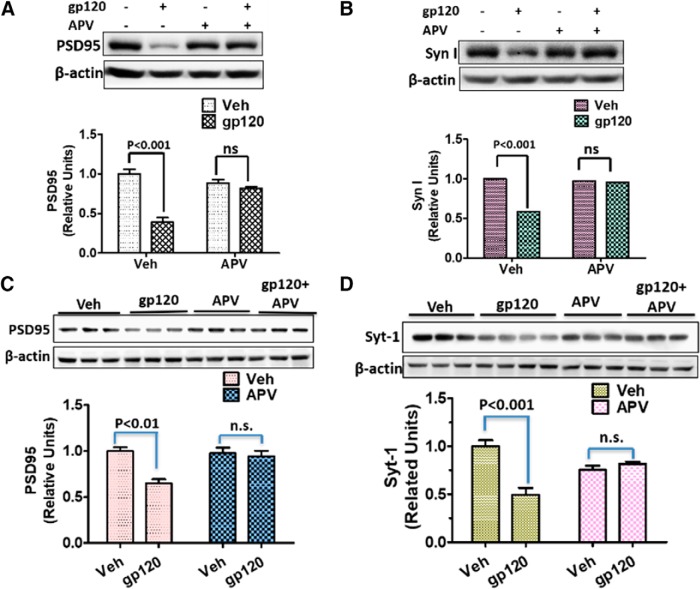
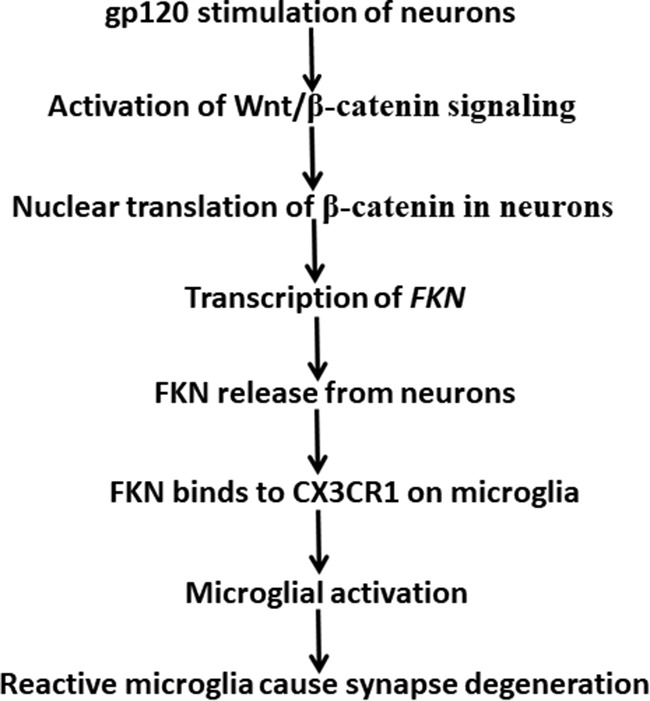
HIV-1 infection of the nervous system causes various neurological diseases, and synaptic degeneration is likely a critical step in the neuropathogenesis. Our prior studies revealed a significant decrease of synaptic protein, specifically in the spinal dorsal horn of patients with HIV-1 in whom pain developed, suggesting a potential contribution of synaptic degeneration to the pathogenesis of HIV-associated pain. However, the mechanism by which HIV-1 causes the spinal synaptic degeneration is unclear. Here, we identified a critical role of microglia in the synaptic degeneration. In primary cortical cultures (day in vitro 14) and spinal cords of 3- to 5-month-old mice (both sexes), microglial ablation inhibited gp120-induced synapse decrease. Fractalkine (FKN), a microglia activation chemokine specifically expressed in neurons, was upregulated by gp120, and knockout of the FKN receptor CX3CR1, which is predominantly expressed in microglia, protected synapses from gp120-induced toxicity. These results indicate that the neuron-to-microglia intercellular FKN/CX3CR1 signaling plays a role in gp120-induced synaptic degeneration. To elucidate the mechanism controlling this intercellular signaling, we tested the role of the Wnt/β-catenin pathway in regulating FKN expression. Inhibition of Wnt/β-catenin signaling blocked both gp120-induced FKN upregulation and synaptic degeneration, and gp120 stimulated Wnt/β-catenin-regulated FKN expression via NMDA receptors (NMDARs). Furthermore, NMDAR antagonist APV, Wnt/β-catenin signaling suppressor DKK1, or knockout of CX3CR1 alleviated gp120-induced mechanical allodynia in mice, suggesting a critical contribution of the Wnt/β-catenin/FKN/CX3R1 pathway to gp120-induced pain. These findings collectively suggest that HIV-1 gp120 induces synaptic degeneration in the spinal pain neural circuit by activating microglia via Wnt3a/β-catenin-regulated FKN expression in neurons.SIGNIFICANCE STATEMENT Synaptic degeneration develops in the spinal cord dorsal horn of HIV patients with chronic pain, but the patients without the pain disorder do not show this neuropathology, indicating a pathogenic contribution of the synaptic degeneration to the development of HIV-associated pain. However, the mechanism underlying the synaptic degeneration is unclear. We report here that HIV-1 gp120, a neurotoxic protein that is specifically associated with the manifestation of pain in HIV patients, induces synapse loss via microglia. Further studies elucidate that gp120 activates microglia by stimulating Wnt/β-catenin-regulated fractalkine in neuron. The results demonstrate a critical role of microglia in the pathogenesis of HIV-associated synaptic degeneration in the spinal pain neural circuit.
Keywords: HIV-1; Wnt; microglia; pain; spinal cord; synaptic degeneration.
Copyright © 2019 the authors.
Figures
Similar articles
-
Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice.J Neurochem. 2010 Aug;114(4):1143-57. doi: 10.1111/j.1471-4159.2010.06837.x. Epub 2010 May 28. J Neurochem. 2010. PMID: 20524966
-
HIV-1 gp120 Upregulates Brain-Derived Neurotrophic Factor (BDNF) Expression in BV2 Cells via the Wnt/β-Catenin Signaling Pathway.J Mol Neurosci. 2017 Jun;62(2):199-208. doi: 10.1007/s12031-017-0931-z. Epub 2017 May 30. J Mol Neurosci. 2017. PMID: 28560687 Free PMC article.
-
Wnt/β-catenin signaling regulates brain-derived neurotrophic factor release from spinal microglia to mediate HIV1 gp120-induced neuropathic pain.Mol Pain. 2020 Jan-Dec;16:1744806920922100. doi: 10.1177/1744806920922100. Mol Pain. 2020. PMID: 32354292 Free PMC article.
-
Fractalkine/CX3CR1-Dependent Modulation of Synaptic and Network Plasticity in Health and Disease.Neural Plast. 2023 Jan 4;2023:4637073. doi: 10.1155/2023/4637073. eCollection 2023. Neural Plast. 2023. PMID: 36644710 Free PMC article. Review.
-
Chemokines, neuronal-glial interactions, and central processing of neuropathic pain.Pharmacol Ther. 2010 Apr;126(1):56-68. doi: 10.1016/j.pharmthera.2010.01.002. Epub 2010 Feb 1. Pharmacol Ther. 2010. PMID: 20117131 Free PMC article. Review.
Cited by
-
Exploring viral neuropathic pain: Molecular mechanisms and therapeutic implications.PLoS Pathog. 2024 Aug 8;20(8):e1012397. doi: 10.1371/journal.ppat.1012397. eCollection 2024 Aug. PLoS Pathog. 2024. PMID: 39116040 Free PMC article. Review.
-
Microglia and Wnt Pathways: Prospects for Inflammation in Alzheimer's Disease.Front Aging Neurosci. 2020 May 14;12:110. doi: 10.3389/fnagi.2020.00110. eCollection 2020. Front Aging Neurosci. 2020. PMID: 32477095 Free PMC article. Review.
-
Human microglial models to study host-virus interactions.Exp Neurol. 2023 May;363:114375. doi: 10.1016/j.expneurol.2023.114375. Epub 2023 Mar 11. Exp Neurol. 2023. PMID: 36907350 Free PMC article. Review.
-
Co-receptor signaling in the pathogenesis of neuroHIV.Retrovirology. 2021 Aug 24;18(1):24. doi: 10.1186/s12977-021-00569-x. Retrovirology. 2021. PMID: 34429135 Free PMC article. Review.
-
Expression of Human Immunodeficiency Virus Transactivator of Transcription (HIV-Tat1-86) Protein Alters Nociceptive Processing that is Sensitive to Anti-Oxidant and Anti-Inflammatory Interventions.J Neuroimmune Pharmacol. 2022 Jun;17(1-2):152-164. doi: 10.1007/s11481-021-09985-4. Epub 2021 Feb 22. J Neuroimmune Pharmacol. 2022. PMID: 33619645 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous