

Dent disease: A window into calcium and phosphate transport
- PMID: 31472005
- PMCID: PMC6815805
- DOI: 10.1111/jcmm.14590
Dent disease: A window into calcium and phosphate transport
Abstract
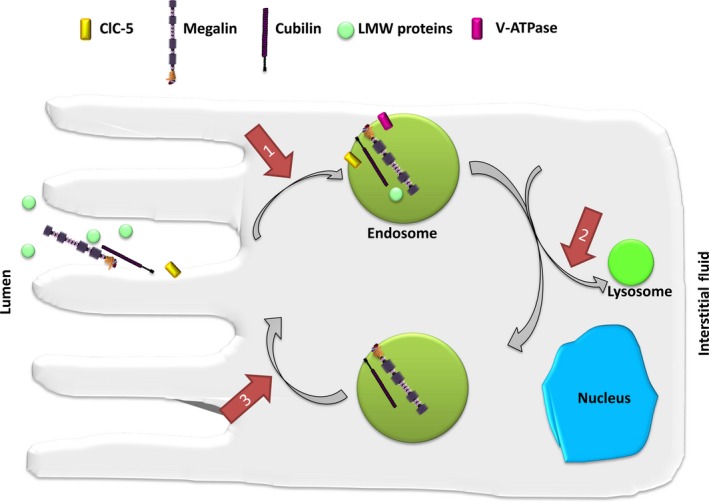
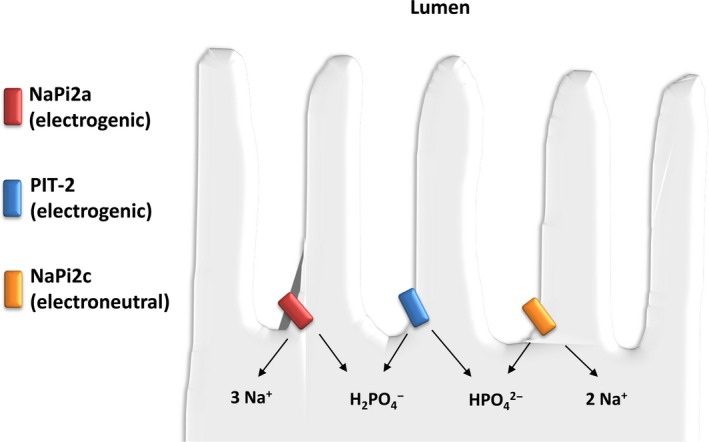
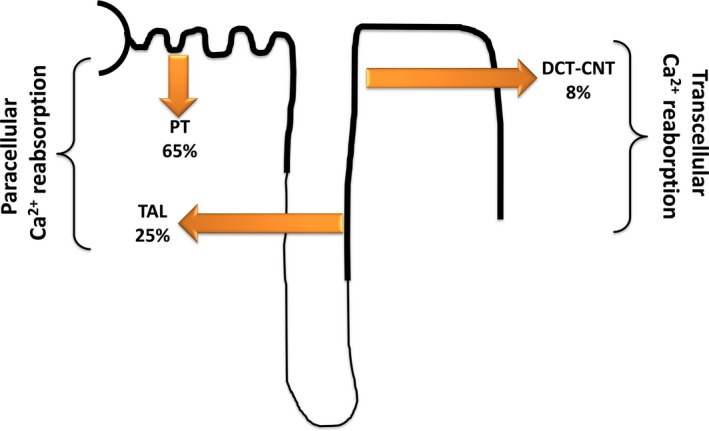
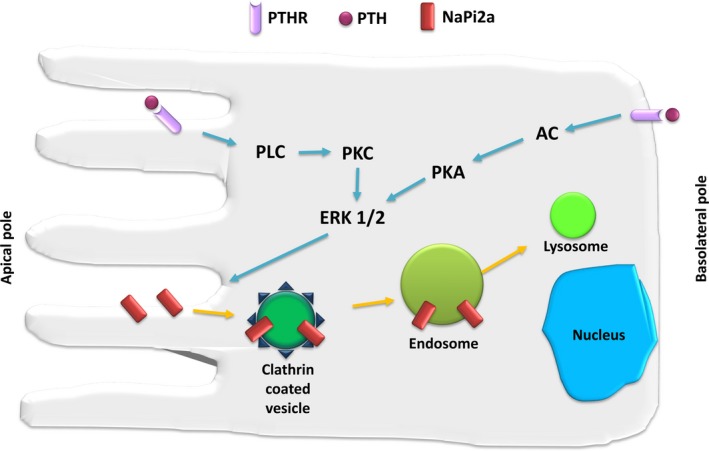
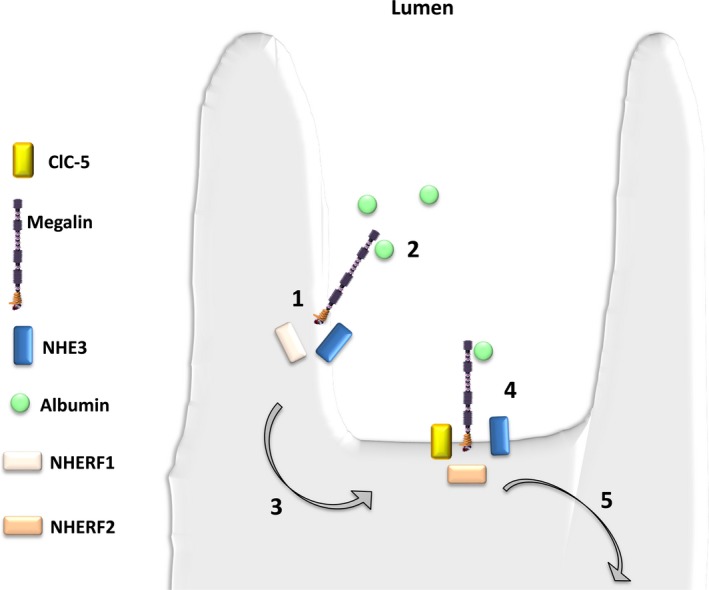
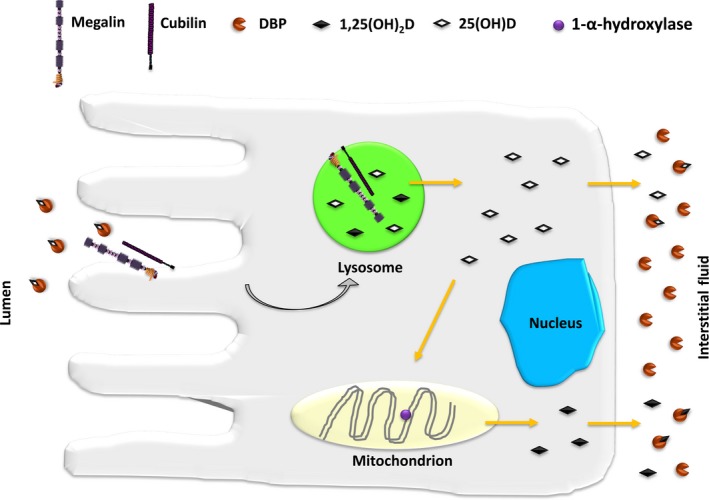
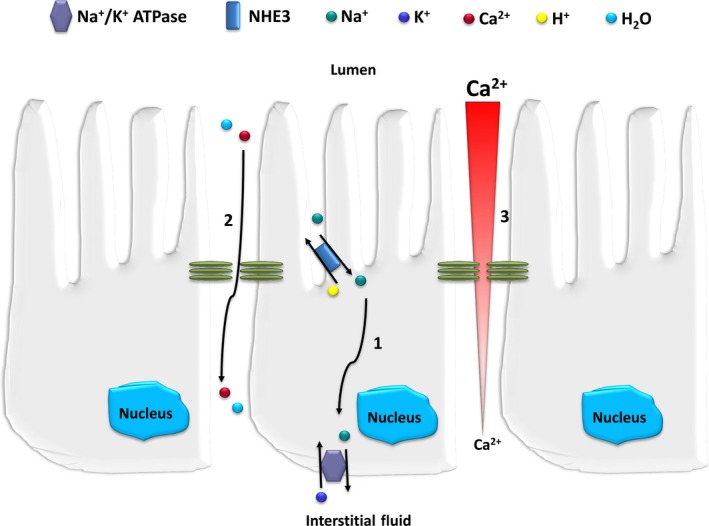
This review examines calcium and phosphate transport in the kidney through the lens of the rare X-linked genetic disorder Dent disease. Dent disease type 1 (DD1) is caused by mutations in the CLCN5 gene encoding ClC-5, a Cl- /H+ antiporter localized to early endosomes of the proximal tubule (PT). Phenotypic features commonly include low molecular weight proteinuria (LMWP), hypercalciuria, focal global sclerosis and chronic kidney disease; calcium nephrolithiasis, nephrocalcinosis and hypophosphatemic rickets are less commonly observed. Although it is not surprising that abnormal endosomal function and recycling in the PT could result in LMWP, it is less clear how ClC-5 dysfunction disturbs calcium and phosphate metabolism. It is known that the majority of calcium and phosphate transport occurs in PT cells, and PT endocytosis is essential for calcium and phosphorus reabsorption in this nephron segment. Evidence from ClC-5 KO models suggests that ClC-5 mediates parathormone endocytosis from tubular fluid. In addition, ClC-5 dysfunction alters expression of the sodium/proton exchanger NHE3 on the PT apical surface thus altering transcellular sodium movement and hence paracellular calcium reabsorption. A potential role for NHE3 dysfunction in the DD1 phenotype has never been investigated, either in DD models or in patients with DD1, even though patients with DD1 exhibit renal sodium and potassium wasting, especially when exposed to even a low dose of thiazide diuretic. Thus, insights from the rare disease DD1 may inform possible underlying mechanisms for the phenotype of hypercalciuria and idiopathic calcium stones.
Keywords: ClC-5; Dent disease; endocytosis; renal calcium transport; renal phosphate transport; sodium/proton exchanger NHE3.
© 2019 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
Conflict of interest statement
The authors confirm that there are no conflicts of interest.
Figures
References
-
- Scheinman SJ. X‐linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int. 1998;53:3‐17. - PubMed
-
- Hoopes RR Jr, Hueber PA, Reid RJ Jr, et al. CLCN5 chloride‐channel mutations in six new North American families with X‐linked nephrolithiasis. Kidney Int. 1998;54:698‐705. - PubMed
-
- Blanchard A, Curis E, Guyon‐Roger T, et al. Observations of a large Dent disease cohort. Kidney Int. 2016;90:430‐439. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
