

Sex-specific changes in the aphid DNA methylation landscape
- PMID: 31472081
- PMCID: PMC6857007
- DOI: 10.1111/mec.15216
Sex-specific changes in the aphid DNA methylation landscape
Abstract
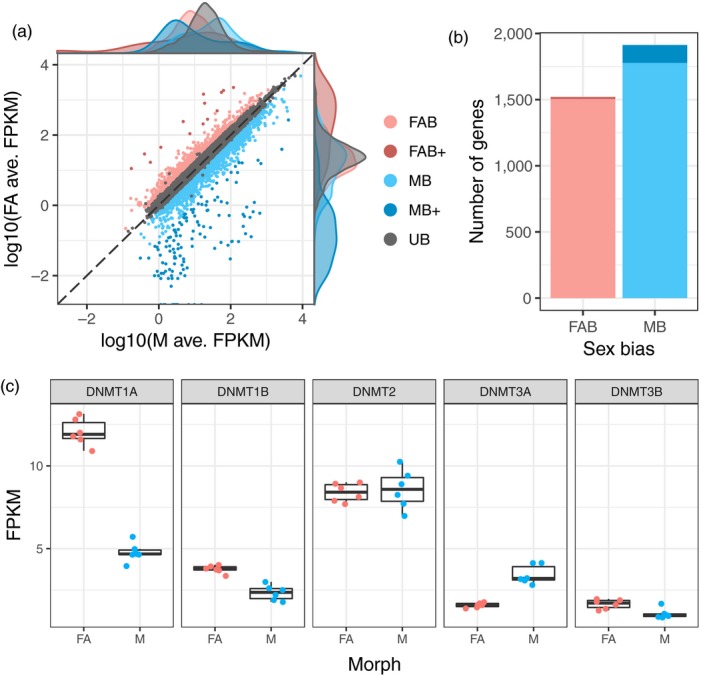
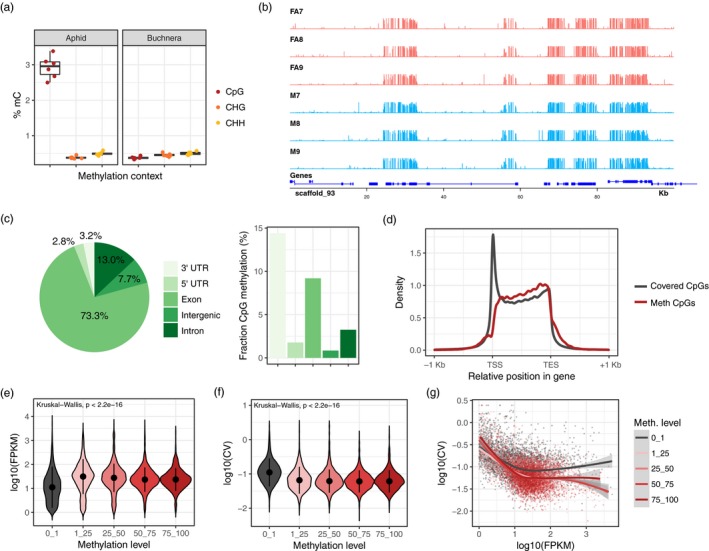
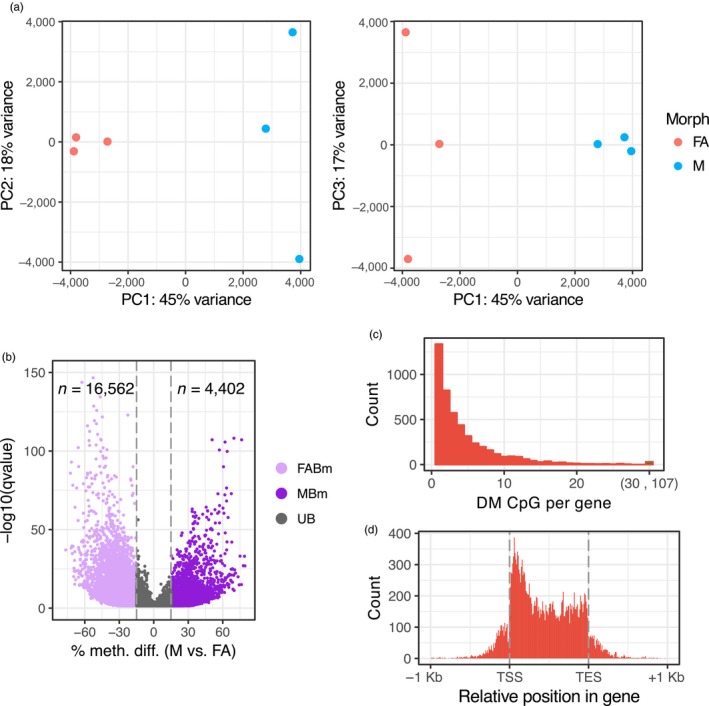
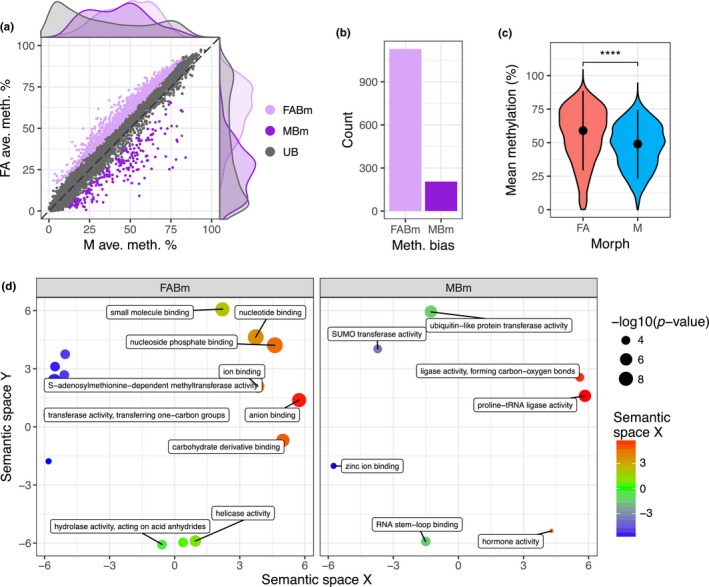
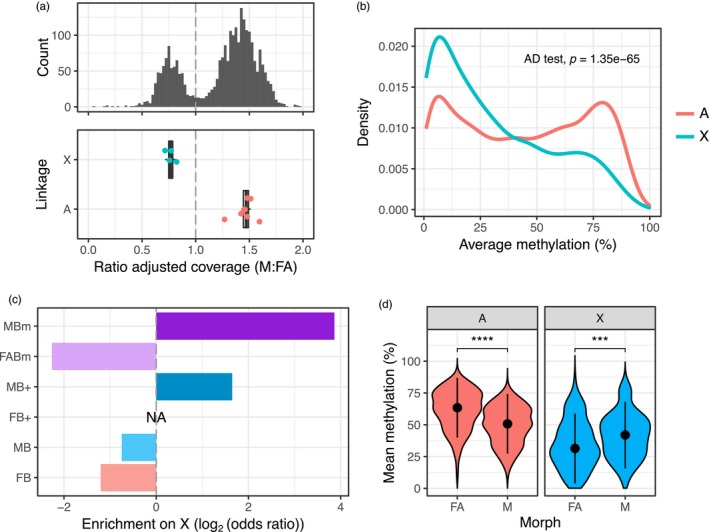
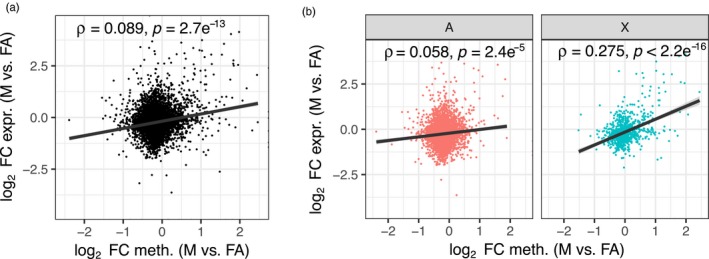
Aphids present an ideal system to study epigenetics as they can produce diverse, but genetically identical, morphs in response to environmental stimuli. Here, using whole genome bisulphite sequencing and transcriptome sequencing of the green peach aphid (Myzus persicae), we present the first detailed analysis of cytosine methylation in an aphid and investigate differences in the methylation and transcriptional landscapes of male and asexual female morphs. We found that methylation primarily occurs in a CG dinucleotide (CpG) context and that exons are highly enriched for methylated CpGs, particularly at the 3' end of genes. Methylation is positively associated with gene expression, and methylated genes are more stably expressed than unmethylated genes. Male and asexual female morphs have distinct methylation profiles. Strikingly, these profiles are divergent between the sex chromosome and the autosomes; autosomal genes are hypomethylated in males compared to asexual females, whereas genes belonging to the sex chromosome, which is haploid in males, are hypermethylated. Overall, we found correlated changes in methylation and gene expression between males and asexual females, and this correlation was particularly strong for genes located on the sex chromosome. Our results suggest that differential methylation of sex-biased genes plays a role in aphid sexual differentiation.
Keywords: Myzus persicae; dosage compensation; epigenetic regulation; sex chromosome; sex-biased gene expression.
© 2019 The Authors. Molecular Ecology published by John Wiley & Sons Ltd.
Figures
References
-
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
- BB/CCG1720/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/L002108/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/N02317X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/P012574/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
