

Ischemic Neuroprotectant PKCε Restores Mitochondrial Glutamate Oxaloacetate Transaminase in the Neuronal NADH Shuttle after Ischemic Injury
- PMID: 31473978
- PMCID: PMC7048657
- DOI: 10.1007/s12975-019-00729-4
Ischemic Neuroprotectant PKCε Restores Mitochondrial Glutamate Oxaloacetate Transaminase in the Neuronal NADH Shuttle after Ischemic Injury
Abstract
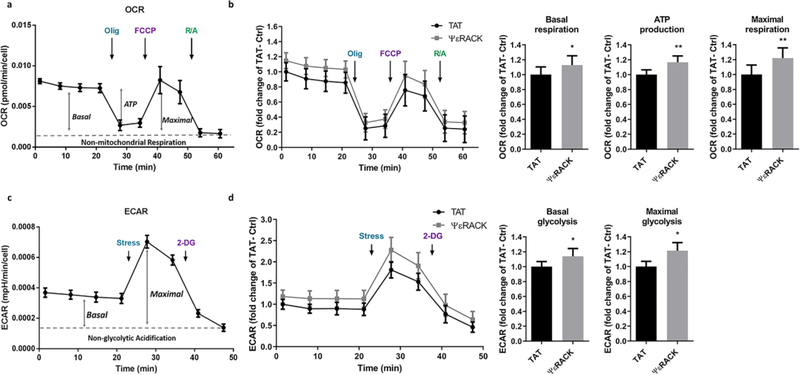
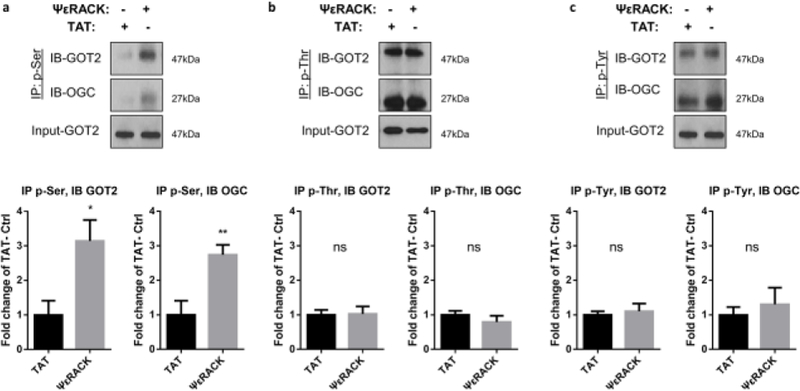
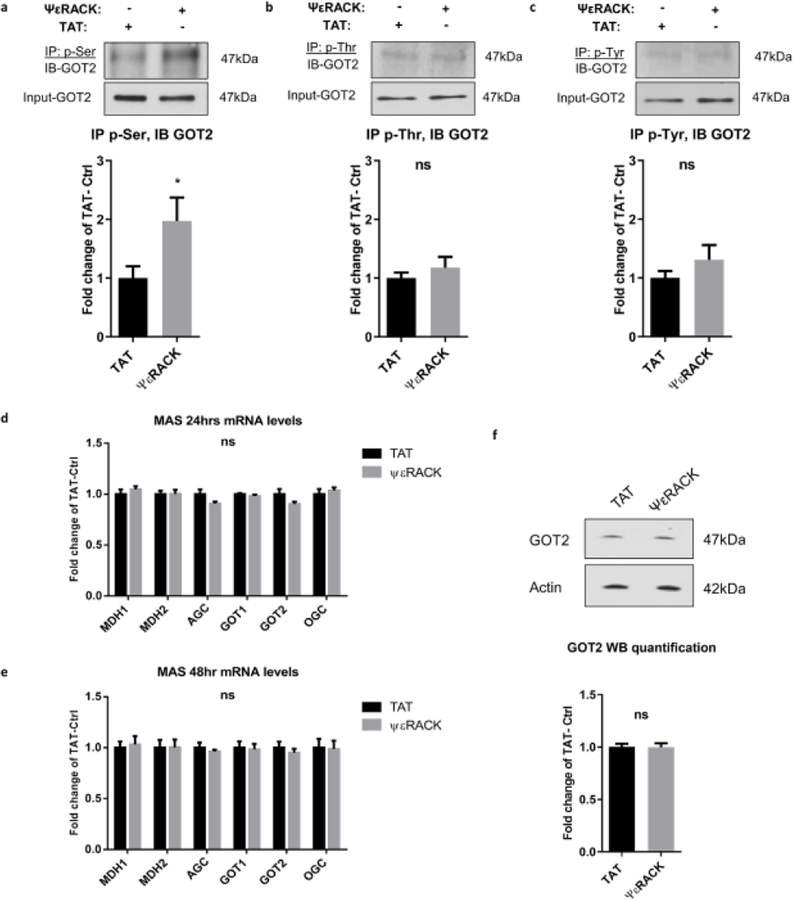
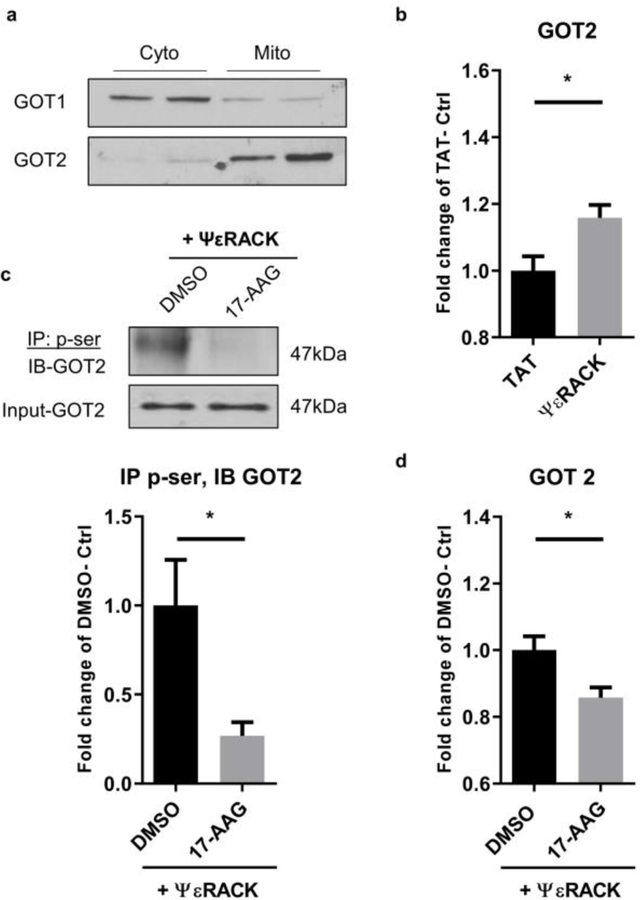
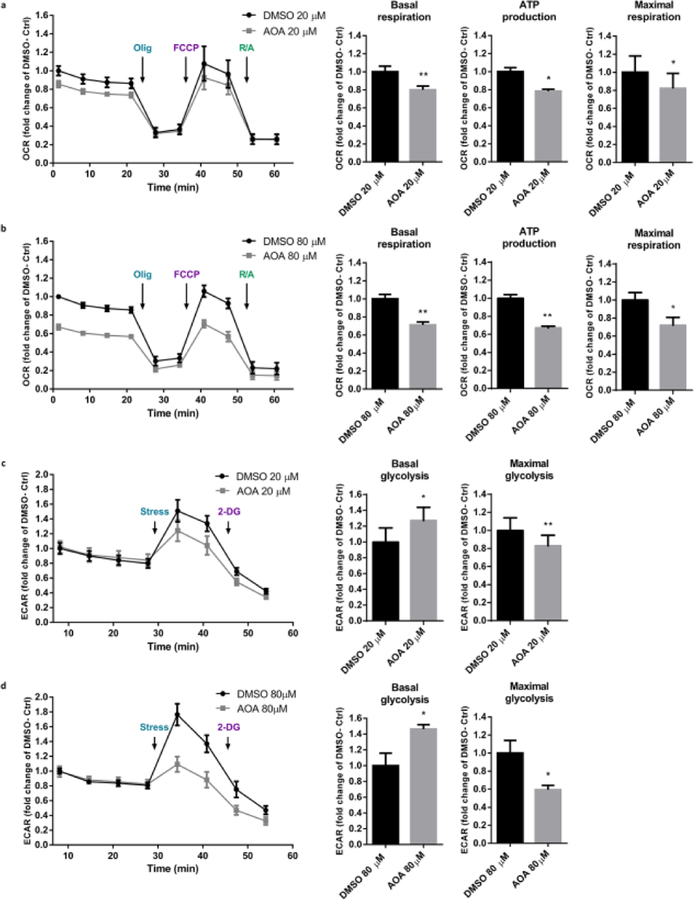
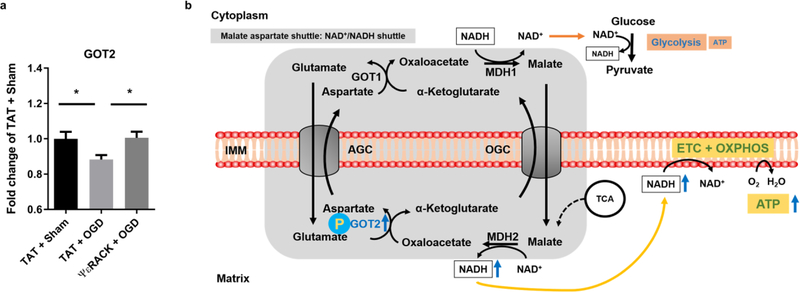
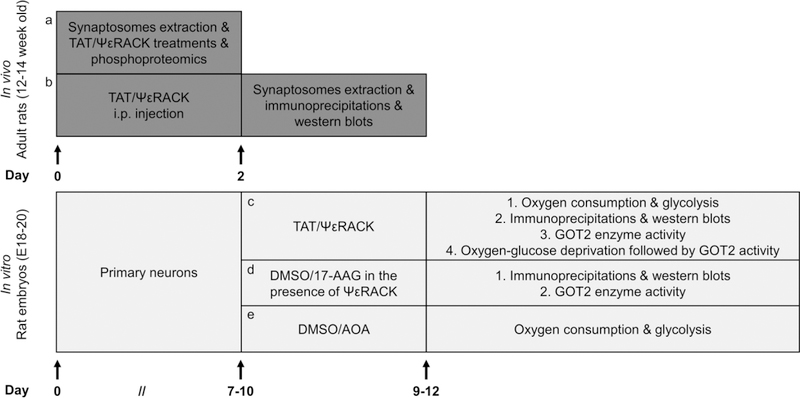
The preservation of mitochondrial function is a major protective strategy for cerebral ischemic injuries. Previously, our laboratory demonstrated that protein kinase C epsilon (PKCε) promotes the synthesis of mitochondrial nicotinamide adenine dinucleotide (NAD+). NAD+ along with its reducing equivalent, NADH, is an essential co-factor needed for energy production from glycolysis and oxidative phosphorylation. Yet, NAD+/NADH are impermeable to the inner mitochondrial membrane and their import into the mitochondria requires the activity of specific shuttles. The most important neuronal NAD+/NADH shuttle is the malate-aspartate shuttle (MAS). The MAS has been implicated in synaptic function and is potentially dysregulated during cerebral ischemia. The aim of this study was to determine if metabolic changes induced by PKCε preconditioning involved regulation of the MAS. Using primary neuronal cultures, we observed that the activation of PKCε enhanced mitochondrial respiration and glycolysis in vitro. Conversely, inhibition of the MAS resulted in decreased oxidative phosphorylation and glycolytic capacity. We further demonstrated that activation of PKCε increased the phosphorylation of key components of the MAS in rat brain synaptosomal fractions. Additionally, PKCε increased the enzyme activity of glutamic oxaloacetic transaminase 2 (GOT2), an effect that was dependent on the import of PKCε into the mitochondria and phosphorylation of GOT2. Furthermore, PKCε activation was able to rescue decreased GOT2 activity induced by ischemia. These findings reveal novel protective targets and mechanisms against ischemic injury, which involves PKCε-mediated phosphorylation and activation of GOT2 in the MAS.
Keywords: Cerebral ischemia; Conditioning; Ischemia tolerance; Ischemic preconditioning; Malate aspartate shuttle; Mitochondria.
Conflict of interest statement
Conflict of Interest
The authors declare that they have no conflict of interest.
The authors declare no potential conflicts of interest.
Figures
References
-
- Kristin V. Ischemic conditioning in organ transplantation. Cond Med. 2018;1(4):212–9.
-
- Cuomo Ornella, Vinciguerra Antonio, Cepparulo Pasquale, Anzilotti Serenella, Brancaccio Paola, Formisano Luigi et al. Differences and similarities in neuroprotective molecular pathways activated by distinct preconditioning inducers. Cond Med. 2018;1(4):187–203.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
