

Germline 16p11.2 Microdeletion Predisposes to Neuroblastoma
- PMID: 31474320
- PMCID: PMC6731370
- DOI: 10.1016/j.ajhg.2019.07.020
Germline 16p11.2 Microdeletion Predisposes to Neuroblastoma
Abstract
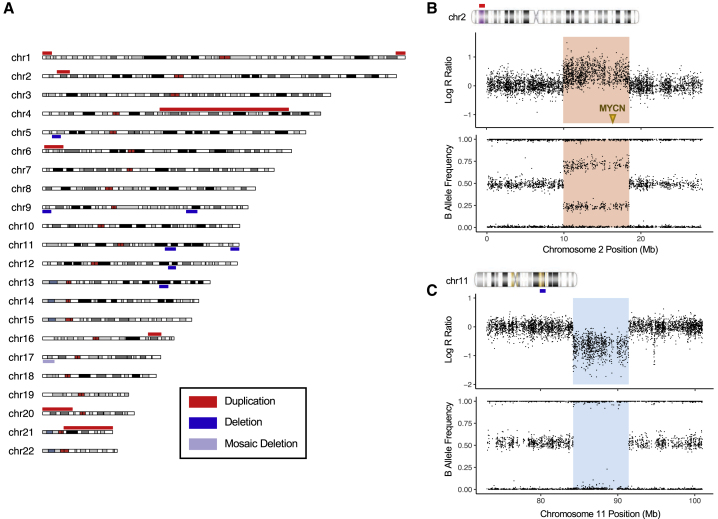
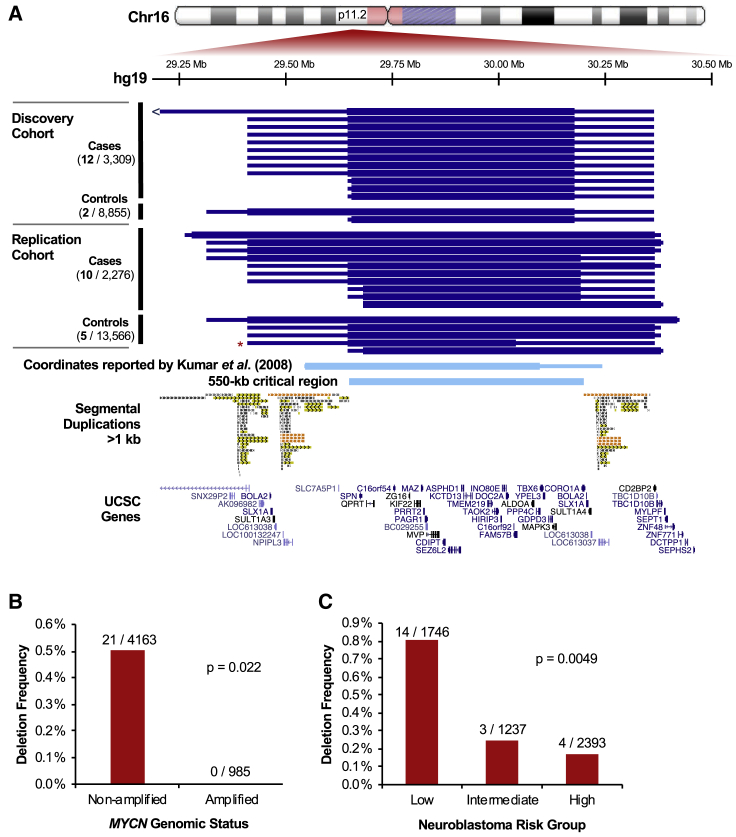
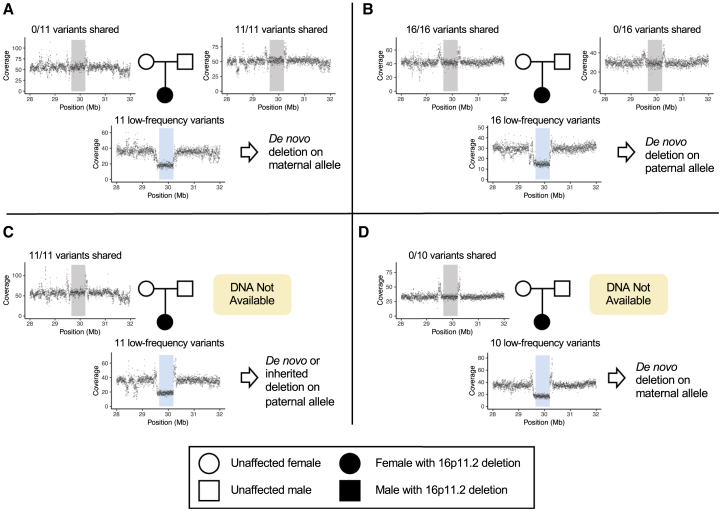
Neuroblastoma is a cancer of the developing sympathetic nervous system. It is diagnosed in 600-700 children per year in the United States and accounts for 12% of pediatric cancer deaths. Despite recent advances in our understanding of this malignancy's complex genetic architecture, the contribution of rare germline variants remains undefined. Here, we conducted a genome-wide analysis of large (>500 kb), rare (<1%) germline copy number variants (CNVs) in two independent, multi-ethnic cohorts totaling 5,585 children with neuroblastoma and 23,505 cancer-free control children. We identified a 550-kb deletion on chromosome 16p11.2 significantly enriched in neuroblastoma cases (0.39% of cases and 0.03% of controls; p = 3.34 × 10-9). Notably, this CNV corresponds to a known microdeletion syndrome that affects approximately one in 3,000 children and confers risk for diverse developmental phenotypes including autism spectrum disorder and other neurodevelopmental disorders. The CNV had a substantial impact on neuroblastoma risk, with an odds ratio of 13.9 (95% confidence interval = 5.8-33.4). The association remained significant when we restricted our analysis to individuals of European ancestry in order to mitigate potential confounding by population stratification (0.42% of cases and 0.03% of controls; p = 4.10 × 10-8). We used whole-genome sequencing (WGS) to validate the deletion in paired germline and tumor DNA from 12 cases. Finally, WGS of four parent-child trios revealed that the deletion primarily arose de novo without maternal or paternal bias. This finding expands the clinical phenotypes associated with 16p11.2 microdeletion syndrome to include cancer, and it suggests that disruption of the 16p11.2 region may dysregulate neurodevelopmental pathways that influence both neurological phenotypes and neuroblastoma.
Keywords: 16p11.2 microdeletion; chromosomal abnormalities; copy number variation; de novo; genetic predisposition; genome-wide association study; germline; neuroblastoma; pediatric cancer; rare variant.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
