

VIVA (VIsualization of VAriants): A VCF File Visualization Tool
- PMID: 31477778
- PMCID: PMC6718772
- DOI: 10.1038/s41598-019-49114-z
VIVA (VIsualization of VAriants): A VCF File Visualization Tool
Abstract
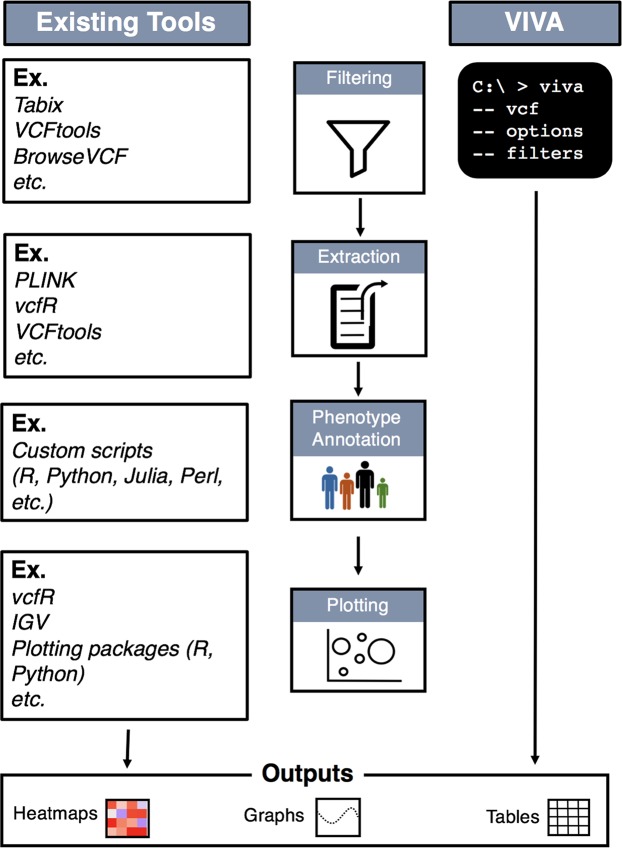
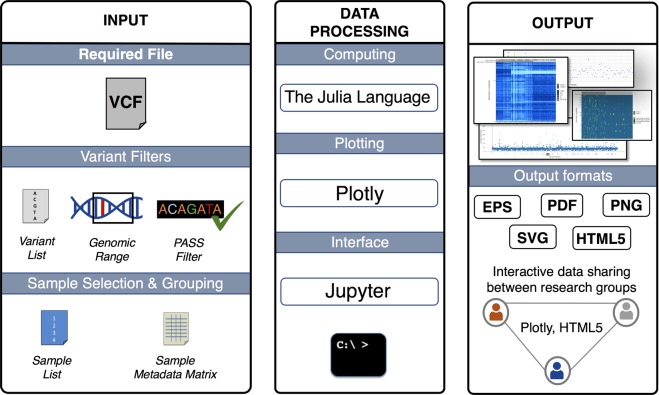
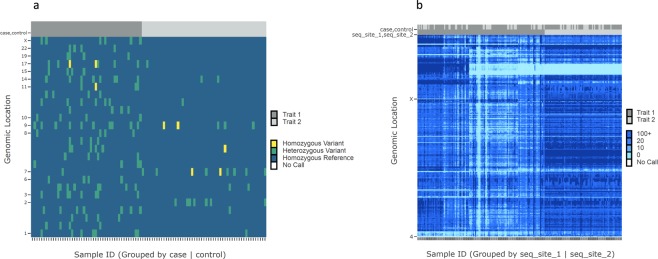
High-throughput sequencing produces an extraordinary amount of genomic data that is organized into a number of high-dimension datasets. Accordingly, visualization of genomic data has become essential for quality control, exploration, and data interpretation. The Variant Call Format (VCF) is a text file format generated during the variant calling process that contains genomic information and locations of variants in a group of sequenced samples. The current workflow for visualization of genomic variant data from VCF files requires use of a combination of existing tools. Here, we describe VIVA (VIsualization of VAriants), a command line utility and Jupyter Notebook based tool for evaluating and sharing genomic data for variant analysis and quality control of sequencing experiments from VCF files. VIVA combines the functionality of existing tools into a single command to interactively evaluate and share genomic data, as well as create publication quality graphics.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
