

Constructing Human Proteoform Families Using Intact-Mass and Top-Down Proteomics with a Multi-Protease Global Post-Translational Modification Discovery Database
- PMID: 31479276
- PMCID: PMC6890418
- DOI: 10.1021/acs.jproteome.9b00339
Constructing Human Proteoform Families Using Intact-Mass and Top-Down Proteomics with a Multi-Protease Global Post-Translational Modification Discovery Database
Abstract
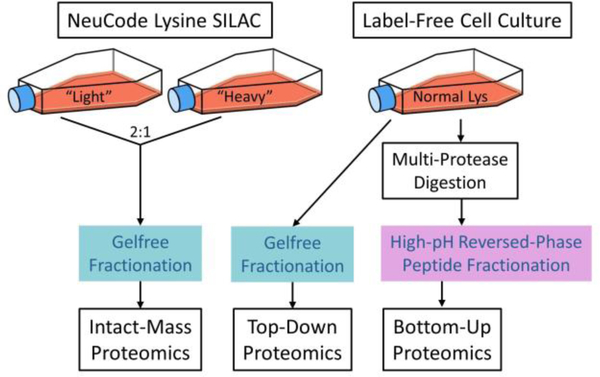
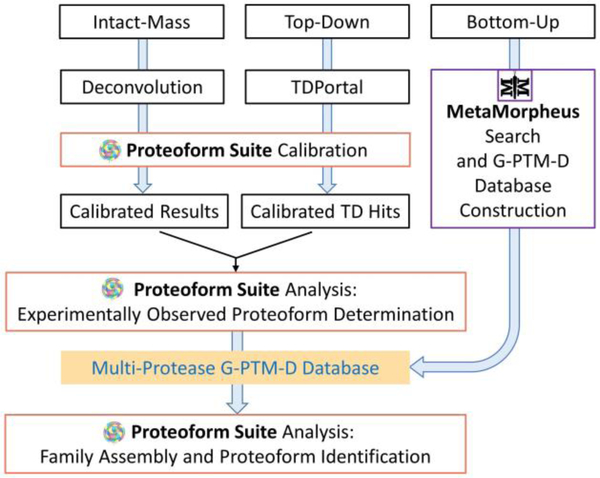
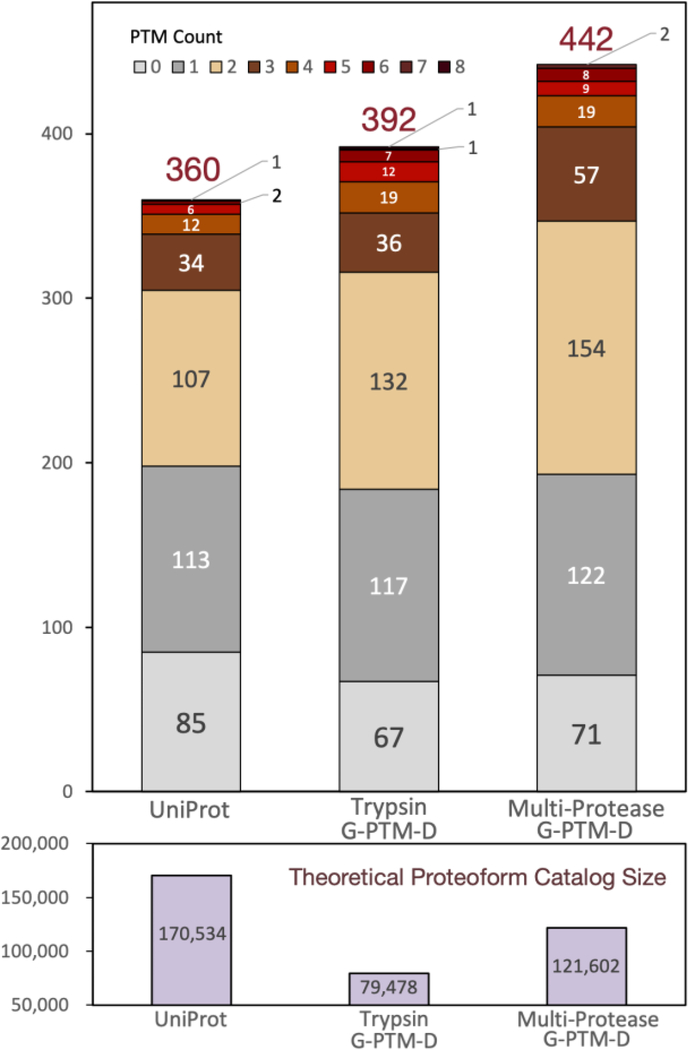
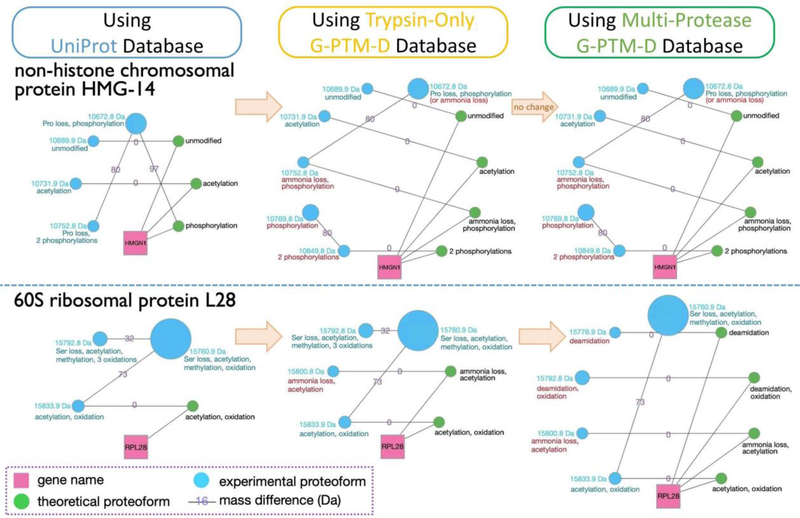
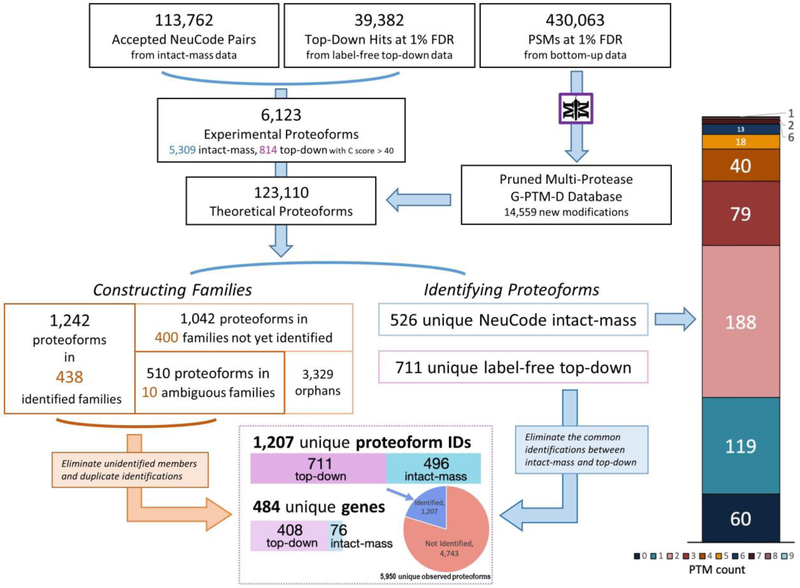
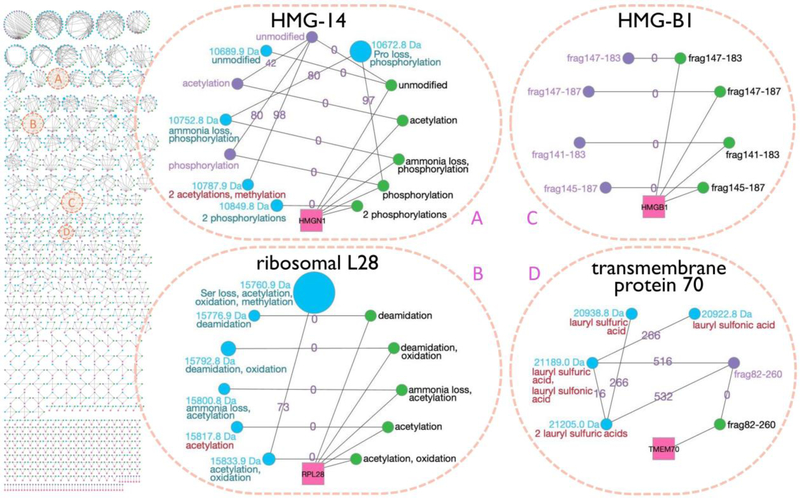
Complex human biomolecular processes are made possible by the diversity of human proteoforms. Constructing proteoform families, groups of proteoforms derived from the same gene, is one way to represent this diversity. Comprehensive, high-confidence identification of human proteoforms remains a central challenge in mass spectrometry-based proteomics. We have previously reported a strategy for proteoform identification using intact-mass measurements, and we have since improved that strategy by mass calibration based on search results, the use of a global post-translational modification discovery database, and the integration of top-down proteomics results with intact-mass analysis. In the present study, we combine these strategies for enhanced proteoform identification in total cell lysate from the Jurkat human T lymphocyte cell line. We collected, processed, and integrated three types of proteomics data (NeuCode-labeled intact-mass, label-free top-down, and multi-protease bottom-up) to maximize the number of confident proteoform identifications. The integrated analysis revealed 5950 unique experimentally observed proteoforms, which were assembled into 848 proteoform families. Twenty percent of the observed proteoforms were confidently identified at a 3.9% false discovery rate, representing 1207 unique proteoforms derived from 484 genes.
Keywords: Jurkat; MetaMorpheus; NeuCode; Proteoform Suite; global PTM discovery; human proteoform; intact-mass; multi-protease; proteoform family; top-down.
Figures
References
-
- Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B; How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
