

Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy
- PMID: 31481971
- PMCID: PMC6710408
- DOI: 10.3389/fgene.2019.00736
Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy
Abstract
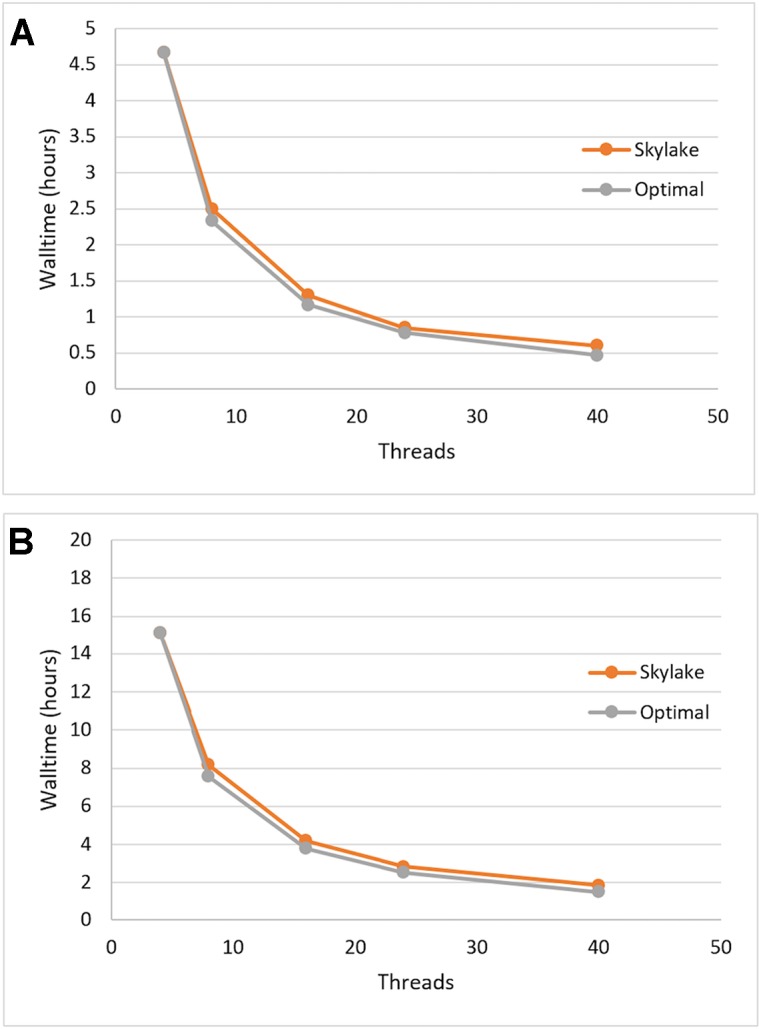
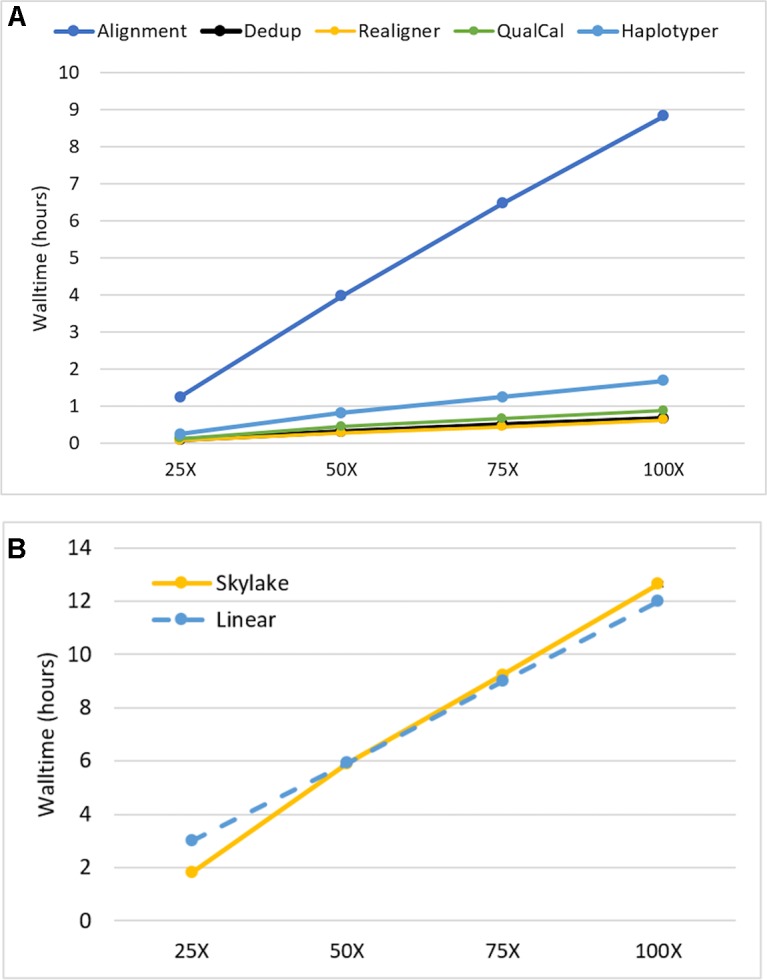
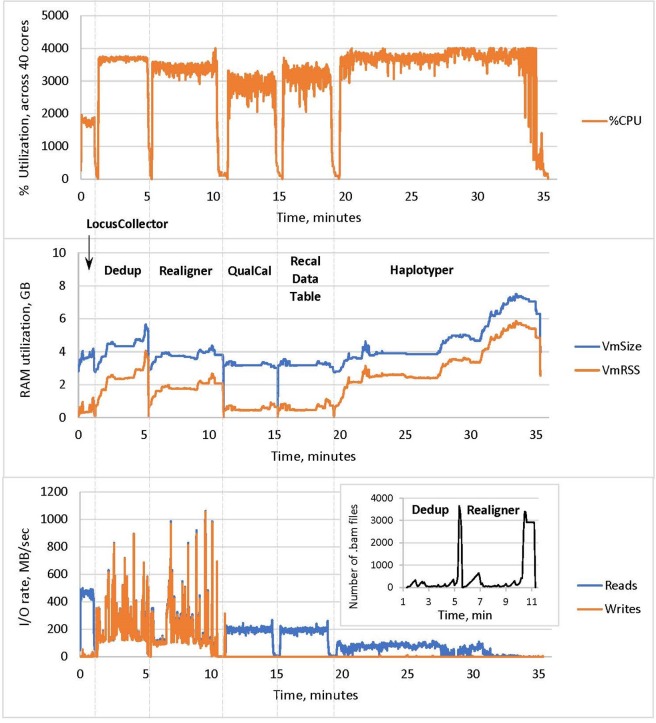
As reliable, efficient genome sequencing becomes ubiquitous, the need for similarly reliable and efficient variant calling becomes increasingly important. The Genome Analysis Toolkit (GATK), maintained by the Broad Institute, is currently the widely accepted standard for variant calling software. However, alternative solutions may provide faster variant calling without sacrificing accuracy. One such alternative is Sentieon DNASeq, a toolkit analogous to GATK but built on a highly optimized backend. We conducted an independent evaluation of the DNASeq single-sample variant calling pipeline in comparison to that of GATK. Our results support the near-identical accuracy of the two software packages, showcase optimal scalability and great speed from Sentieon, and describe computational performance considerations for the deployment of DNASeq.
Keywords: DNASeq; GATK; Sentieon; benchmarking; variant calling.
Figures
References
-
- Banerjee S. S., Athreya A. P., Mainzer L. S., Jongeneel C. V., Hwu W.-M., Kalbarczyk Z. T., et al. (2016). Efficient and scalable workflows for genomic analyses. In Proceedings of the ACM International Workshop on Data-Intensive Distributed Computing (ACM; ), 27–36. 10.1145/2912152.2912156 - DOI
-
- Broad Institute (2018). GATK | Archived versions. https://software.broadinstitute.org/gatk/download/archive.
-
- Chapman B. (2014). Benchmarking variation and rna-seq analyses on amazon web services with docker. Blue Collar Bioinformatics.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials