

Visualizing Proteasome Activity and Intracellular Localization Using Fluorescent Proteins and Activity-Based Probes
- PMID: 31482094
- PMCID: PMC6710370
- DOI: 10.3389/fmolb.2019.00056
Visualizing Proteasome Activity and Intracellular Localization Using Fluorescent Proteins and Activity-Based Probes
Abstract
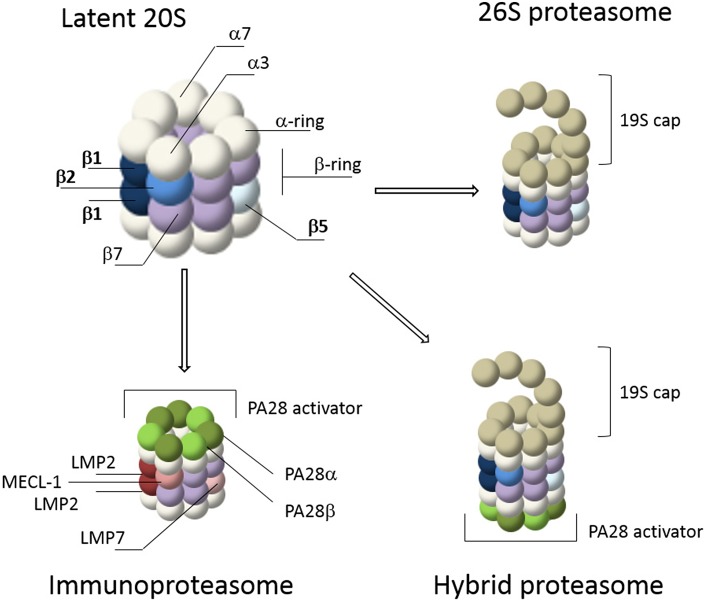
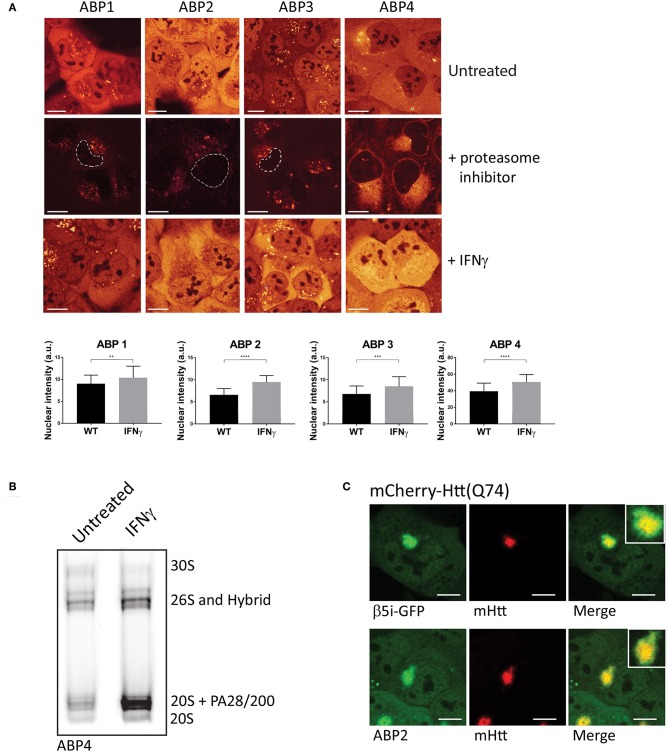
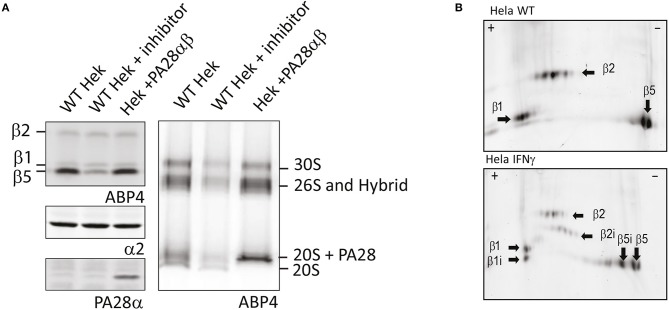
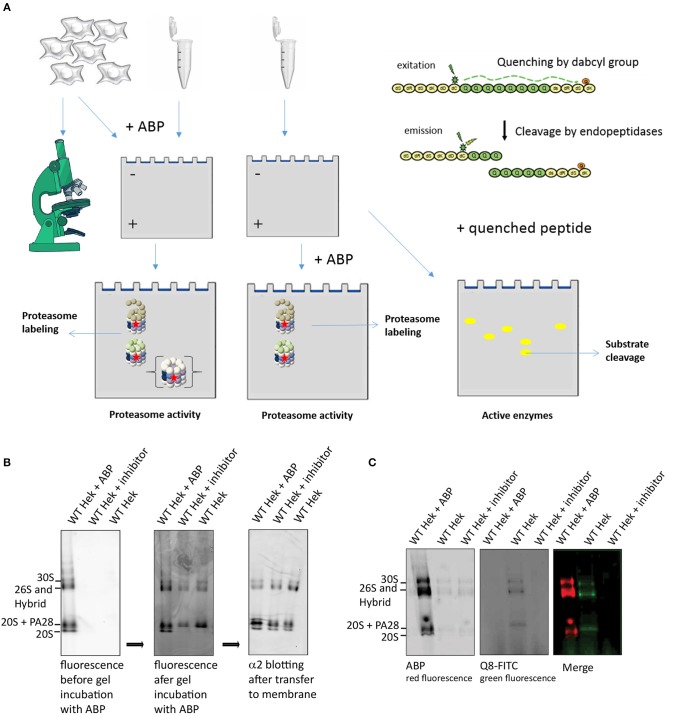
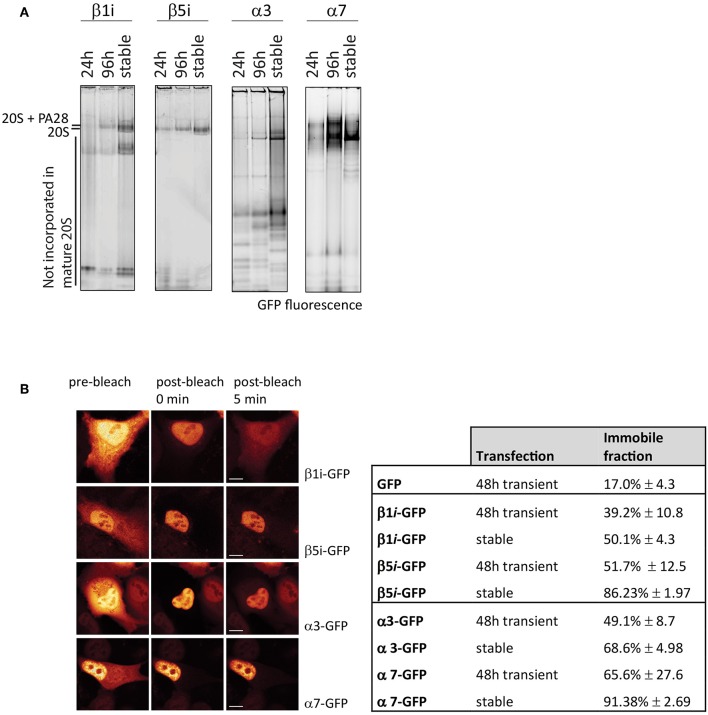
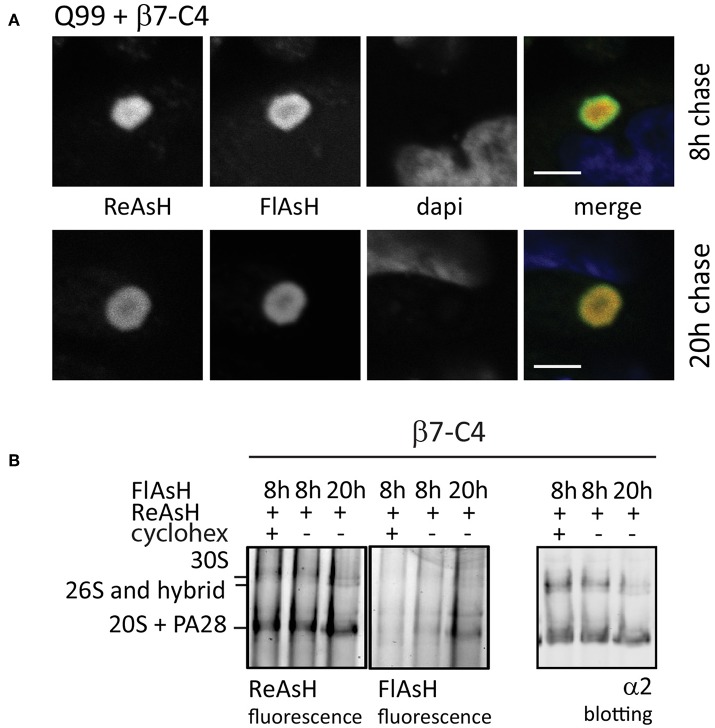
The proteasome is a multi-catalytic molecular machine that plays a key role in the degradation of many cytoplasmic and nuclear proteins. The proteasome is essential and proteasome malfunction is associated with various disease pathologies. Proteasome activity depends on its catalytic subunits which are interchangeable and also on the interaction with the associated regulatory cap complexes. Here, we describe and compare various methods that allow the study of proteasome function in living cells. Methods include the use of fluorescently tagged proteasome subunits and the use of activity-based proteasome probes. These probes can be used in both biochemical assays and in microscopy-based experiments. Together with tagged proteasomes, they can be used to study proteasome localization, dynamics, and activity.
Keywords: activity probes; dynamics; fluorescence; living cells; proteasome.
Figures

References
LinkOut - more resources
Full Text Sources
Miscellaneous