

Relative stabilities of wild-type and mutant glial fibrillary acidic protein in patients with Alexander disease
- PMID: 31484723
- PMCID: PMC6816090
- DOI: 10.1074/jbc.RA119.009777
Relative stabilities of wild-type and mutant glial fibrillary acidic protein in patients with Alexander disease
Abstract
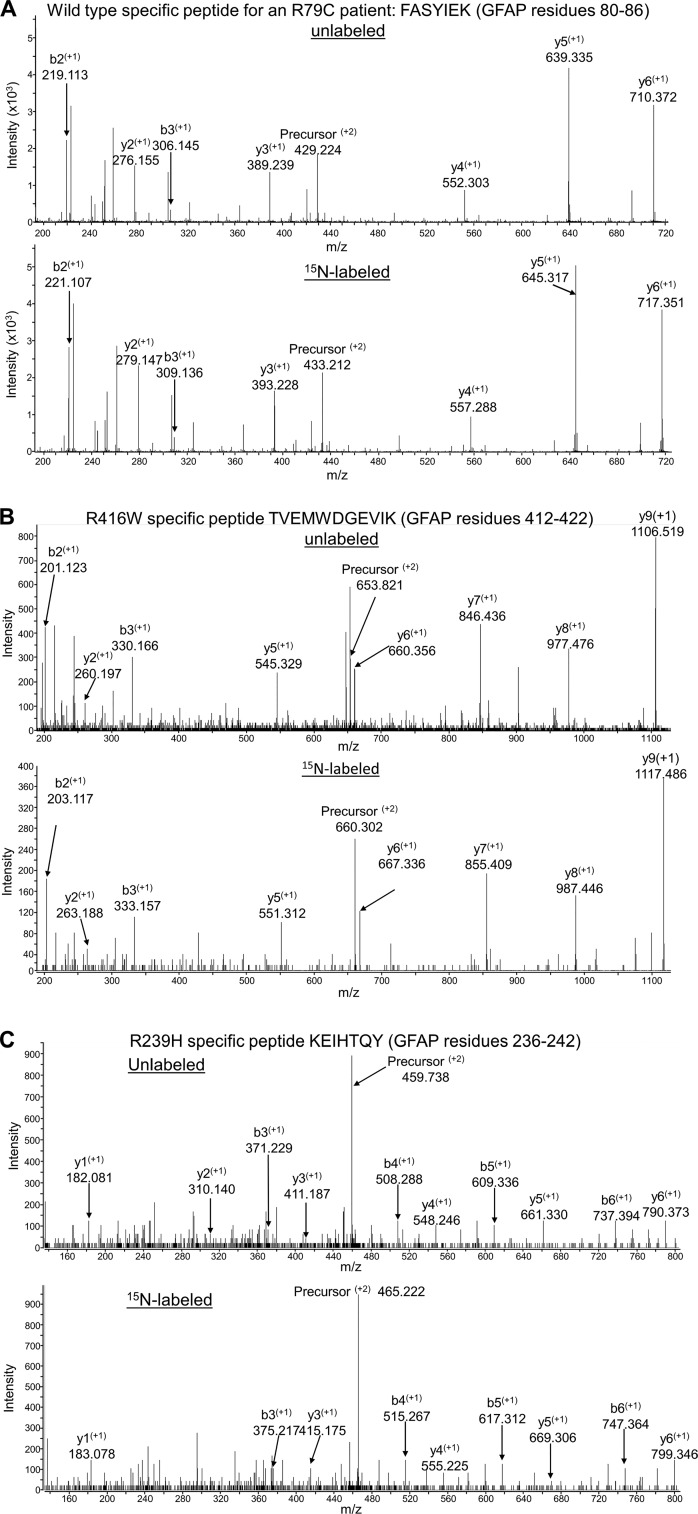
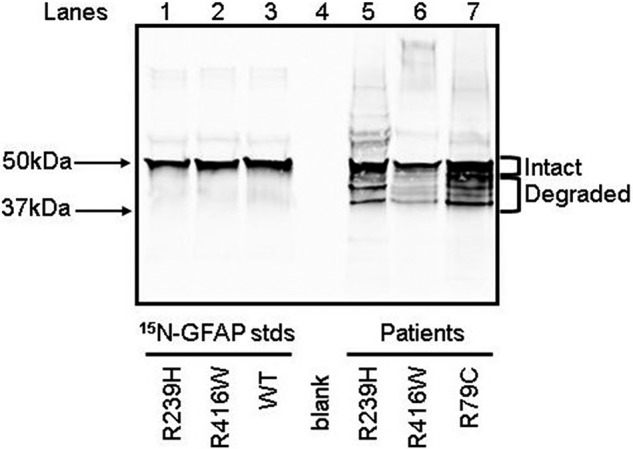
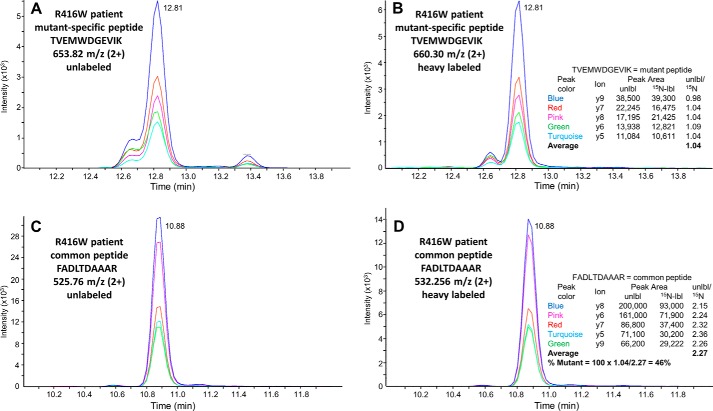
Alexander disease (AxD) is an often fatal astrogliopathy caused by dominant gain-of-function missense mutations in the glial fibrillary acidic protein (GFAP) gene. The mechanism by which the mutations produce the AxD phenotype is not known. However, the observation that features of AxD are displayed by mice that express elevated levels of GFAP from a human WT GFAP transgene has contributed to the notion that the mutations produce AxD by increasing accumulation of total GFAP above some toxic threshold rather than the mutant GFAP being inherently toxic. A possible mechanism for accumulation of GFAP in AxD patients is that the mutated GFAP variants are more stable than the WT, an attribution abetted by observations that GFAP complexes containing GFAP variants are more resistant to solvent extraction. Here we tested this hypothesis by determining the relative levels of WT and mutant GFAP in three individuals with AxD, each of whom carried a common but different GFAP mutation (R79C, R239H, or R416W). Mass spectrometry analysis identified a peptide specific to the mutant or WT GFAP in each patient, and we quantified this peptide by comparing its signal to that of an added [15N]GFAP standard. In all three individuals, the level of mutant GFAP was less than that of the WT. This finding suggests that AxD onset is due to an intrinsic toxicity of the mutant GFAP instead of it acting indirectly by being more stable than WT GFAP and thereby increasing the total GFAP level.
Keywords: Alexander disease; MS; astrocyte; astrogliopathy; genetic disease; glial fibrillary acidic protein (GFAP); intermediate filament; mutant; protein stability.
© 2019 Heaven et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Figures
Similar articles
-
Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease.J Biol Chem. 2017 Apr 7;292(14):5814-5824. doi: 10.1074/jbc.M116.772020. Epub 2017 Feb 21. J Biol Chem. 2017. PMID: 28223355 Free PMC article.
-
Glial fibrillary acidic protein is pathologically modified in Alexander disease.J Biol Chem. 2024 Jul;300(7):107402. doi: 10.1016/j.jbc.2024.107402. Epub 2024 May 21. J Biol Chem. 2024. PMID: 38782207 Free PMC article.
-
Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease.Am J Pathol. 2006 Mar;168(3):888-97. doi: 10.2353/ajpath.2006.051028. Am J Pathol. 2006. PMID: 16507904 Free PMC article.
-
Myelin changes in Alexander disease.Neurologia (Engl Ed). 2018 Oct;33(8):526-533. doi: 10.1016/j.nrl.2017.01.019. Epub 2017 Mar 22. Neurologia (Engl Ed). 2018. PMID: 28342553 Review. English, Spanish.
-
GFAP mutations in Alexander disease.Int J Dev Neurosci. 2002 Jun-Aug;20(3-5):259-68. doi: 10.1016/s0736-5748(02)00019-9. Int J Dev Neurosci. 2002. PMID: 12175861 Review.
Cited by
-
Alexander disease: models, mechanisms, and medicine.Curr Opin Neurobiol. 2022 Feb;72:140-147. doi: 10.1016/j.conb.2021.10.002. Epub 2021 Nov 23. Curr Opin Neurobiol. 2022. PMID: 34826654 Free PMC article. Review.
-
Roles of the cytoskeleton in human diseases.Mol Biol Rep. 2023 Mar;50(3):2847-2856. doi: 10.1007/s11033-022-08025-5. Epub 2023 Jan 6. Mol Biol Rep. 2023. PMID: 36609753 Review.
-
Alexander disease: the road ahead.Neural Regen Res. 2023 Oct;18(10):2156-2160. doi: 10.4103/1673-5374.369097. Neural Regen Res. 2023. PMID: 37056123 Free PMC article. Review.
-
GFAP at 50.ASN Neuro. 2020 Jan-Dec;12:1759091420949680. doi: 10.1177/1759091420949680. ASN Neuro. 2020. PMID: 32811163 Free PMC article. Review.
-
Effects of Alexander disease-associated mutations on the assembly and organization of GFAP intermediate filaments.Mol Biol Cell. 2022 Jul 1;33(8):ar69. doi: 10.1091/mbc.E22-01-0013. Epub 2022 May 5. Mol Biol Cell. 2022. PMID: 35511821 Free PMC article.
References
-
- Brenner M., Goldman J. E., Quinlan R. A., and Messing A. (2009) in Astrocytes in (Patho)physiology of the Nervous System (Parpura V., and Haydon P., eds.) pp. 591–648, Springer, New York
-
- Flint D., and Brenner M. (2011) in Leukodystrophies (Raymond G. V., Eichler F., Fatemi A., and Naidu S., eds.) pp. 106–129, Mac Keith Press, London
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
