

Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints
- PMID: 31484923
- PMCID: PMC6726615
- DOI: 10.1038/s41467-019-11994-0
Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints
Abstract
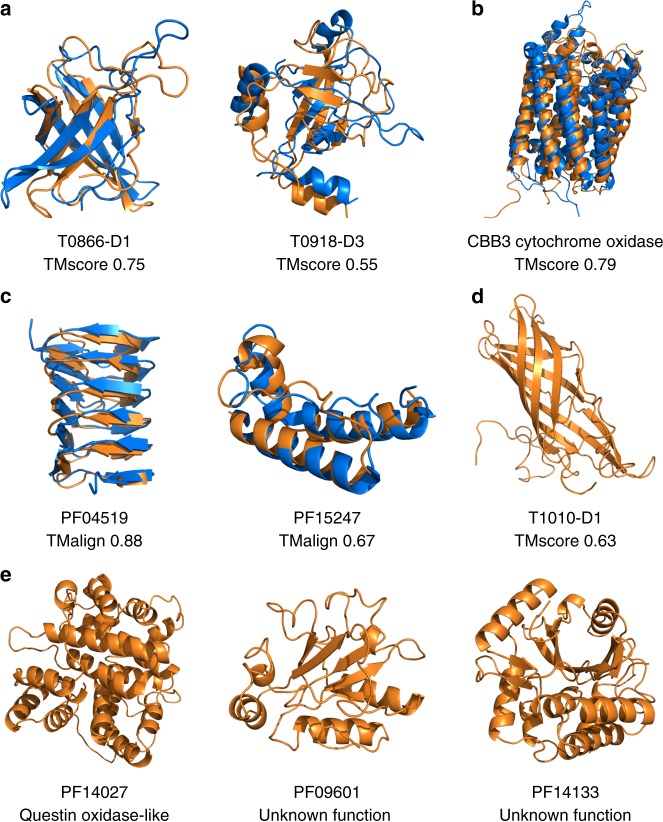
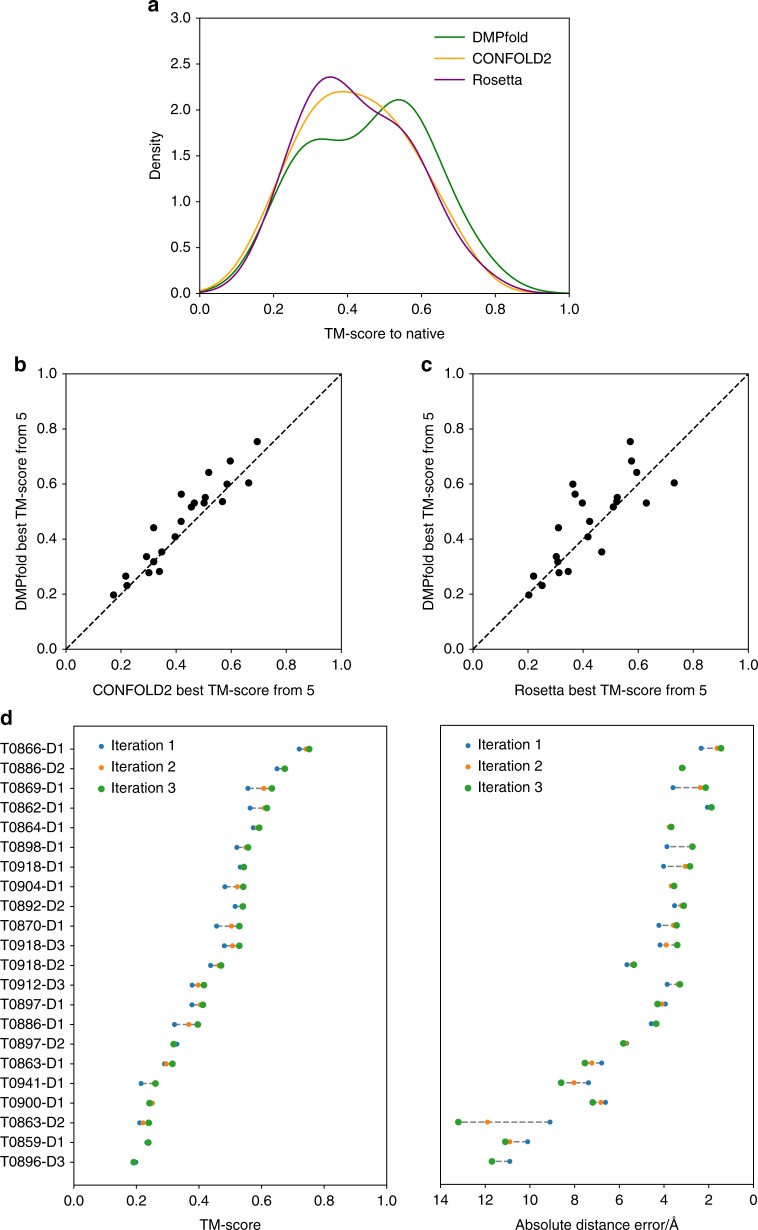
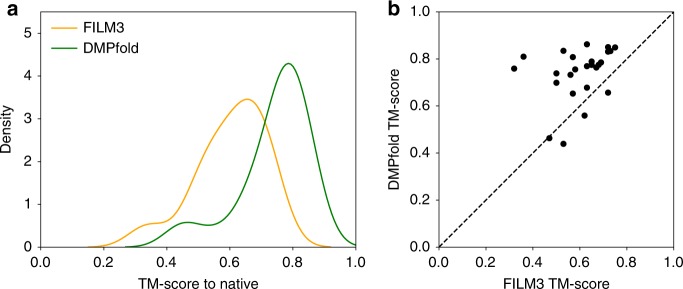
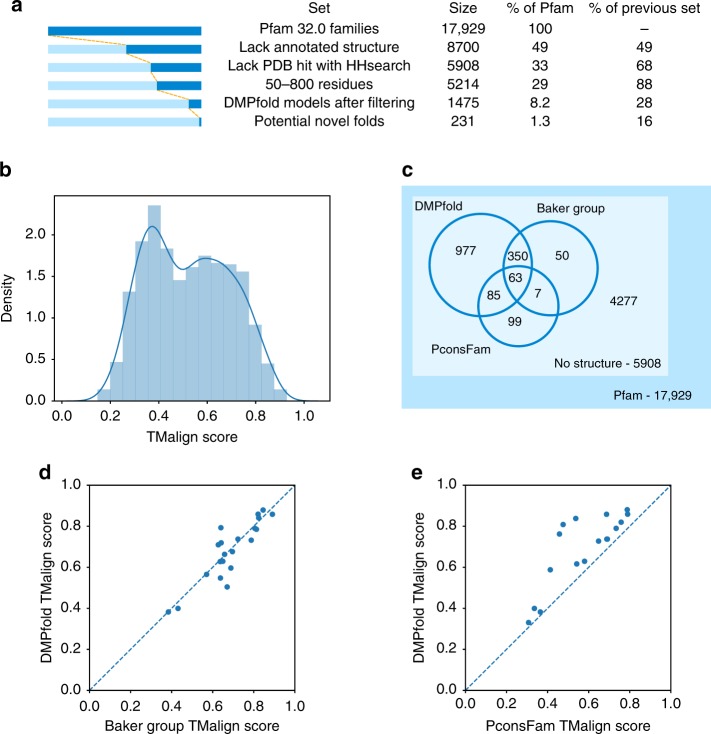
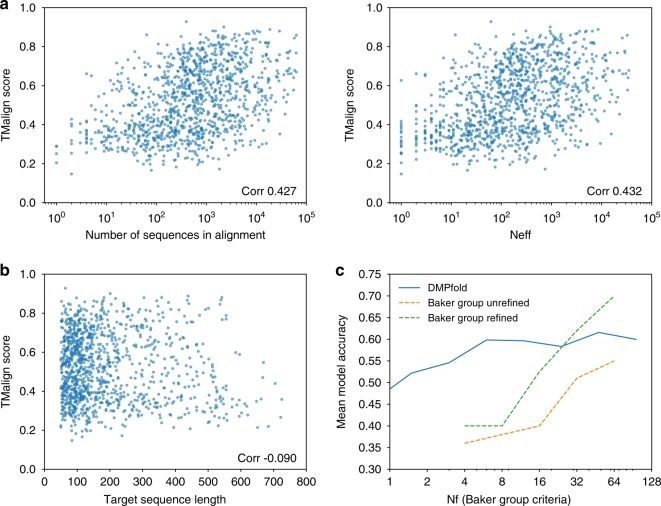
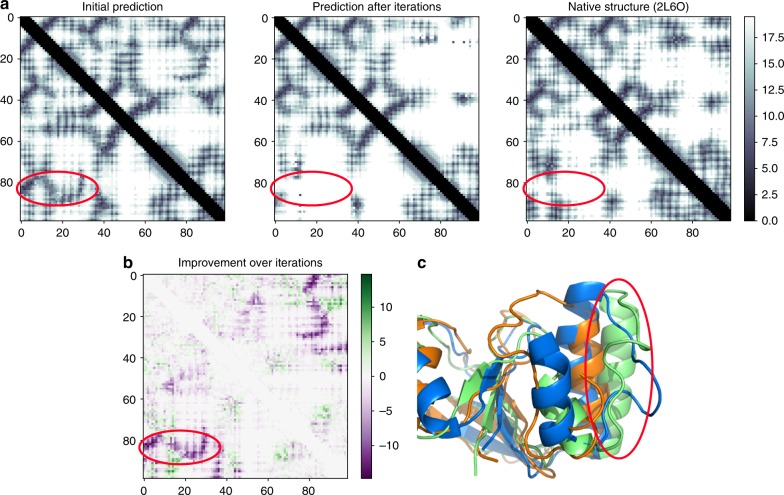
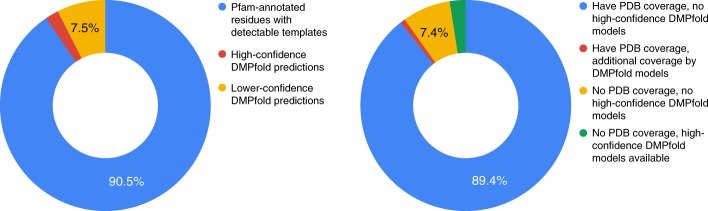
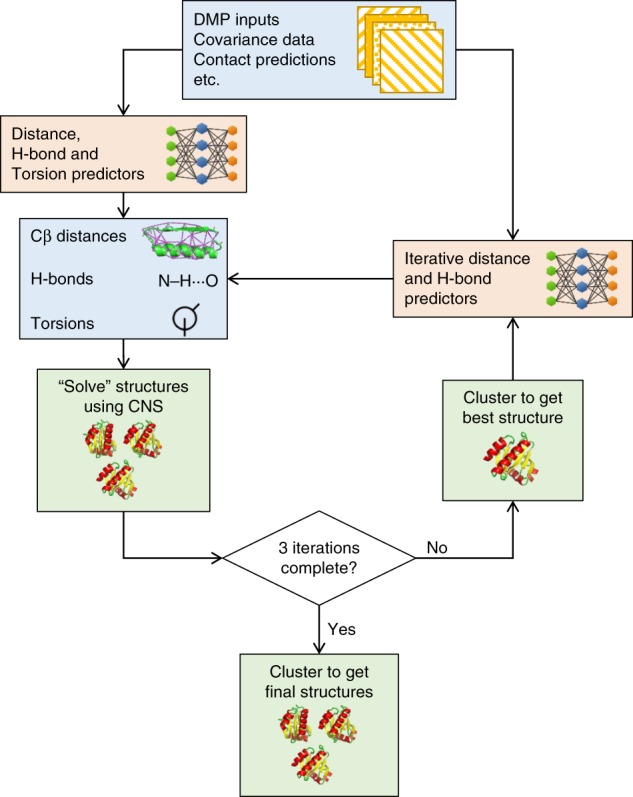
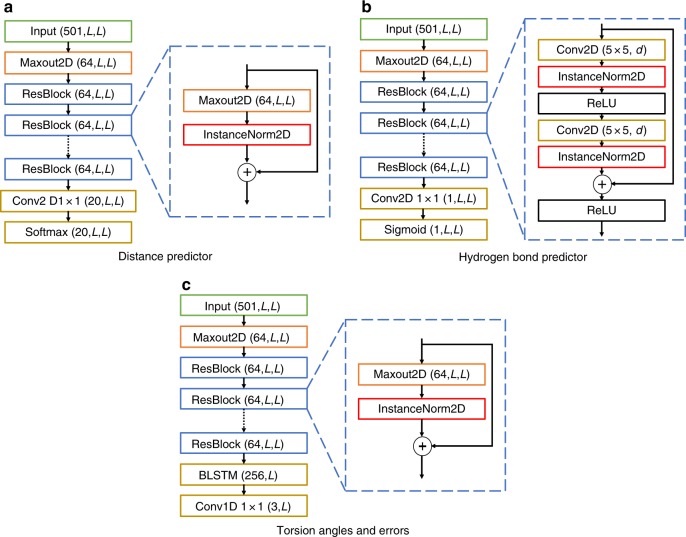
The inapplicability of amino acid covariation methods to small protein families has limited their use for structural annotation of whole genomes. Recently, deep learning has shown promise in allowing accurate residue-residue contact prediction even for shallow sequence alignments. Here we introduce DMPfold, which uses deep learning to predict inter-atomic distance bounds, the main chain hydrogen bond network, and torsion angles, which it uses to build models in an iterative fashion. DMPfold produces more accurate models than two popular methods for a test set of CASP12 domains, and works just as well for transmembrane proteins. Applied to all Pfam domains without known structures, confident models for 25% of these so-called dark families were produced in under a week on a small 200 core cluster. DMPfold provides models for 16% of human proteome UniProt entries without structures, generates accurate models with fewer than 100 sequences in some cases, and is freely available.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Protein contact prediction by integrating deep multiple sequence alignments, coevolution and machine learning.Proteins. 2018 Mar;86 Suppl 1(Suppl 1):84-96. doi: 10.1002/prot.25405. Epub 2017 Oct 31. Proteins. 2018. PMID: 29047157 Free PMC article.
-
DeepCDpred: Inter-residue distance and contact prediction for improved prediction of protein structure.PLoS One. 2019 Jan 8;14(1):e0205214. doi: 10.1371/journal.pone.0205214. eCollection 2019. PLoS One. 2019. PMID: 30620738 Free PMC article.
-
Using deep learning to annotate the protein universe.Nat Biotechnol. 2022 Jun;40(6):932-937. doi: 10.1038/s41587-021-01179-w. Epub 2022 Feb 21. Nat Biotechnol. 2022. PMID: 35190689
-
Deep learning methods for 3D structural proteome and interactome modeling.Curr Opin Struct Biol. 2022 Apr;73:102329. doi: 10.1016/j.sbi.2022.102329. Epub 2022 Feb 6. Curr Opin Struct Biol. 2022. PMID: 35139457 Free PMC article. Review.
-
The AlphaFold Database of Protein Structures: A Biologist's Guide.J Mol Biol. 2022 Jan 30;434(2):167336. doi: 10.1016/j.jmb.2021.167336. Epub 2021 Oct 29. J Mol Biol. 2022. PMID: 34757056 Free PMC article. Review.
Cited by
-
Fast and accurate Ab Initio Protein structure prediction using deep learning potentials.PLoS Comput Biol. 2022 Sep 16;18(9):e1010539. doi: 10.1371/journal.pcbi.1010539. eCollection 2022 Sep. PLoS Comput Biol. 2022. PMID: 36112717 Free PMC article.
-
A Novel Protein from Ectocarpus sp. Improves Salinity and High Temperature Stress Tolerance in Arabidopsis thaliana.Int J Mol Sci. 2021 Feb 17;22(4):1971. doi: 10.3390/ijms22041971. Int J Mol Sci. 2021. PMID: 33671243 Free PMC article.
-
DEMO2: Assemble multi-domain protein structures by coupling analogous template alignments with deep-learning inter-domain restraint prediction.Nucleic Acids Res. 2022 Jul 5;50(W1):W235-W245. doi: 10.1093/nar/gkac340. Nucleic Acids Res. 2022. PMID: 35536281 Free PMC article.
-
A Vaccine Construction against COVID-19-Associated Mucormycosis Contrived with Immunoinformatics-Based Scavenging of Potential Mucoralean Epitopes.Vaccines (Basel). 2022 Apr 22;10(5):664. doi: 10.3390/vaccines10050664. Vaccines (Basel). 2022. PMID: 35632420 Free PMC article.
-
AI in health and medicine.Nat Med. 2022 Jan;28(1):31-38. doi: 10.1038/s41591-021-01614-0. Epub 2022 Jan 20. Nat Med. 2022. PMID: 35058619 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
