

Interrogating Mutant Allele Expression via Customized Reference Genomes to Define Influential Cancer Mutations
- PMID: 31484939
- PMCID: PMC6726654
- DOI: 10.1038/s41598-019-48967-8
Interrogating Mutant Allele Expression via Customized Reference Genomes to Define Influential Cancer Mutations
Abstract
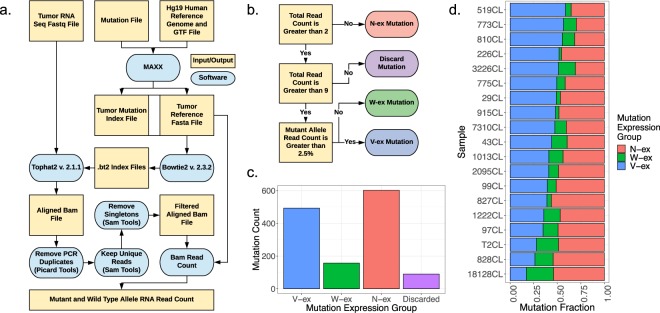
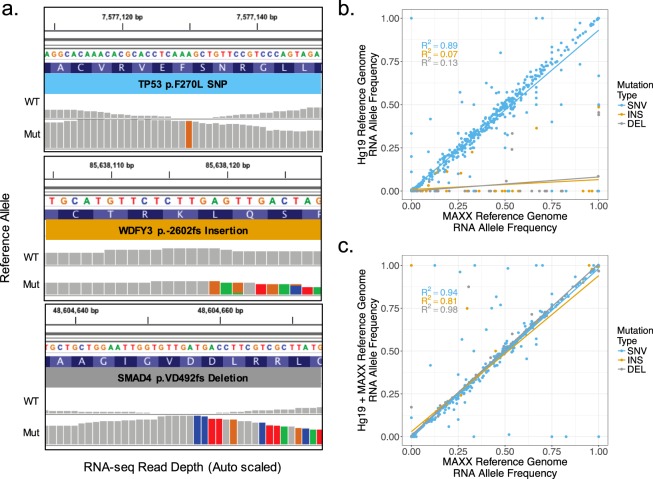
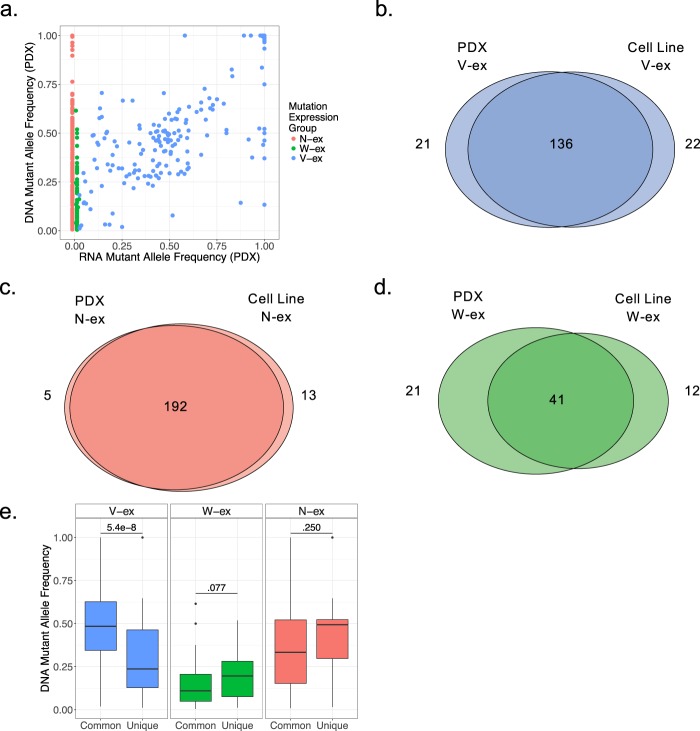
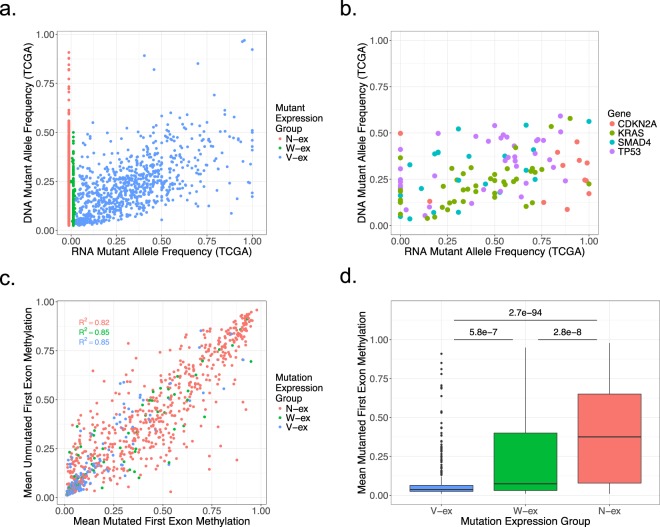
Genetic alterations are essential for cancer initiation and progression. However, differentiating mutations that drive the tumor phenotype from mutations that do not affect tumor fitness remains a fundamental challenge in cancer biology. To better understand the impact of a given mutation within cancer, RNA-sequencing data was used to categorize mutations based on their allelic expression. For this purpose, we developed the MAXX (Mutation Allelic Expression Extractor) software, which is highly effective at delineating the allelic expression of both single nucleotide variants and small insertions and deletions. Results from MAXX demonstrated that mutations can be separated into three groups based on their expression of the mutant allele, lack of expression from both alleles, or expression of only the wild-type allele. By taking into consideration the allelic expression patterns of genes that are mutated in PDAC, it was possible to increase the sensitivity of widely used driver mutation detection methods, as well as identify subtypes that have prognostic significance and are associated with sensitivity to select classes of therapeutic agents in cell culture. Thus, differentiating mutations based on their mutant allele expression via MAXX represents a means to parse somatic variants in tumor genomes, helping to elucidate a gene's respective role in cancer.
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
IDENTIFY CANCER DRIVER GENES THROUGH SHARED MENDELIAN DISEASE PATHOGENIC VARIANTS AND CANCER SOMATIC MUTATIONS.Pac Symp Biocomput. 2017;22:473-484. doi: 10.1142/9789813207813_0044. Pac Symp Biocomput. 2017. PMID: 27896999
-
The search for cis-regulatory driver mutations in cancer genomes.Oncotarget. 2015 Oct 20;6(32):32509-25. doi: 10.18632/oncotarget.5085. Oncotarget. 2015. PMID: 26356674 Free PMC article. Review.
-
Systematic pan-cancer analysis of somatic allele frequency.Sci Rep. 2018 May 16;8(1):7735. doi: 10.1038/s41598-018-25462-0. Sci Rep. 2018. PMID: 29769535 Free PMC article.
-
Exome-wide analysis of bi-allelic alterations identifies a Lynch phenotype in The Cancer Genome Atlas.Genome Med. 2018 Sep 14;10(1):69. doi: 10.1186/s13073-018-0579-5. Genome Med. 2018. PMID: 30217226 Free PMC article.
-
Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes.Brief Bioinform. 2016 Jul;17(4):642-56. doi: 10.1093/bib/bbv068. Epub 2015 Aug 24. Brief Bioinform. 2016. PMID: 26307061 Free PMC article. Review.
Cited by
-
Maximizing Small Biopsy Patient Samples: Unified RNA-Seq Platform Assessment of over 120,000 Patient Biopsies.J Pers Med. 2022 Dec 22;13(1):24. doi: 10.3390/jpm13010024. J Pers Med. 2022. PMID: 36675685 Free PMC article.
-
Quantification of mutant-allele expression at isoform level in cancer from RNA-seq data.NAR Genom Bioinform. 2022 Jul 13;4(3):lqac052. doi: 10.1093/nargab/lqac052. eCollection 2022 Sep. NAR Genom Bioinform. 2022. PMID: 35855322 Free PMC article.
-
RCCC_Pred: A Novel Method for Sequence-Based Identification of Renal Clear Cell Carcinoma Genes through DNA Mutations and a Blend of Features.Diagnostics (Basel). 2022 Dec 3;12(12):3036. doi: 10.3390/diagnostics12123036. Diagnostics (Basel). 2022. PMID: 36553042 Free PMC article.
-
Identification and Validation of T-cell Receptors Targeting RAS Hotspot Mutations in Human Cancers for Use in Cell-based Immunotherapy.Clin Cancer Res. 2021 Sep 15;27(18):5084-5095. doi: 10.1158/1078-0432.CCR-21-0849. Epub 2021 Jun 24. Clin Cancer Res. 2021. PMID: 34168045 Free PMC article.
-
Cancer Neoantigens: Challenges and Future Directions for Prediction, Prioritization, and Validation.Front Oncol. 2022 Mar 3;12:836821. doi: 10.3389/fonc.2022.836821. eCollection 2022. Front Oncol. 2022. PMID: 35311072 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
