

REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification
- PMID: 31484976
- PMCID: PMC6726608
- DOI: 10.1038/s41598-019-49224-8
REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification
Abstract
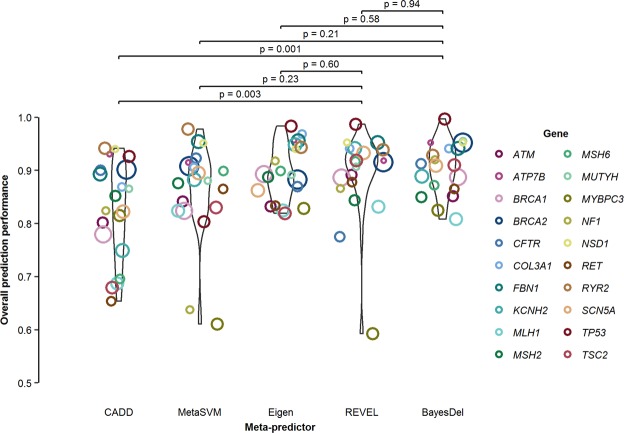
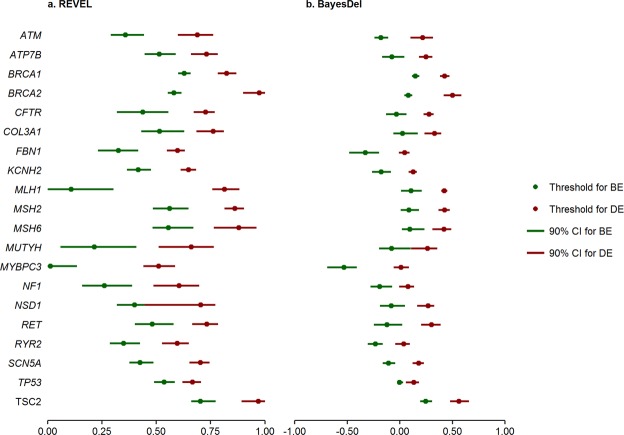
Many in silico predictors of genetic variant pathogenicity have been previously developed, but there is currently no standard application of these algorithms for variant assessment. Using 4,094 ClinVar-curated missense variants in clinically actionable genes, we evaluated the accuracy and yield of benign and deleterious evidence in 5 in silico meta-predictors, as well as agreement of SIFT and PolyPhen2, and report the derived thresholds for the best performing predictor(s). REVEL and BayesDel outperformed all other meta-predictors (CADD, MetaSVM, Eigen), with higher positive predictive value, comparable negative predictive value, higher yield, and greater overall prediction performance. Agreement of SIFT and PolyPhen2 resulted in slightly higher yield but lower overall prediction performance than REVEL or BayesDel. Our results support the use of gene-level rather than generalized thresholds, when gene-level thresholds can be estimated. Our results also support the use of 2-sided thresholds, which allow for uncertainty, rather than a single, binary cut-point for assigning benign and deleterious evidence. The gene-level 2-sided thresholds we derived for REVEL or BayesDel can be used to assess in silico evidence for missense variants in accordance with current classification guidelines.
Conflict of interest statement
All of the authors are employed by and receive a salary from Ambry Genetics.
Figures
References
LinkOut - more resources
Full Text Sources
