

The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape
- PMID: 31485078
- PMCID: PMC6742567
- DOI: 10.1038/s41586-019-1534-3
The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape
Abstract
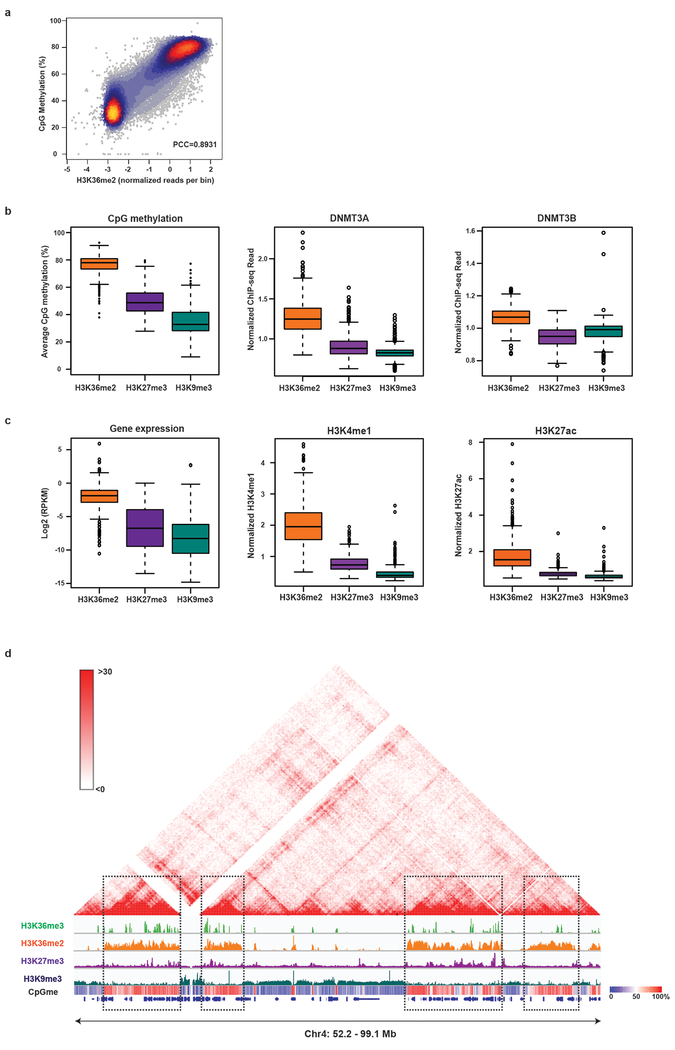
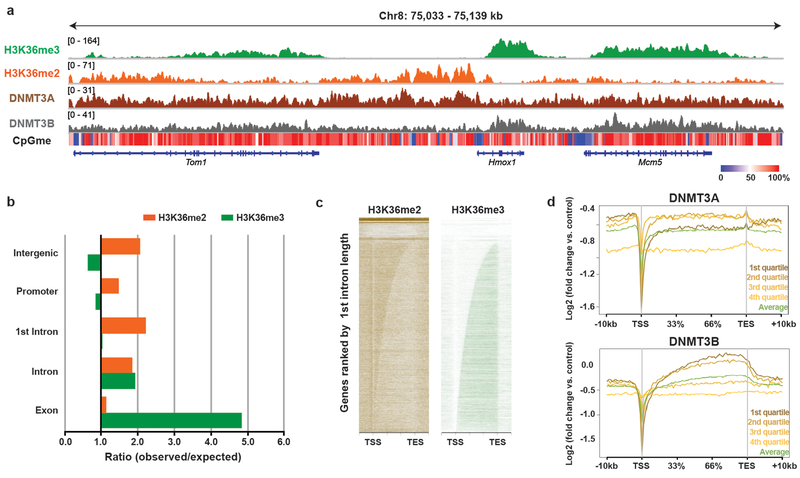
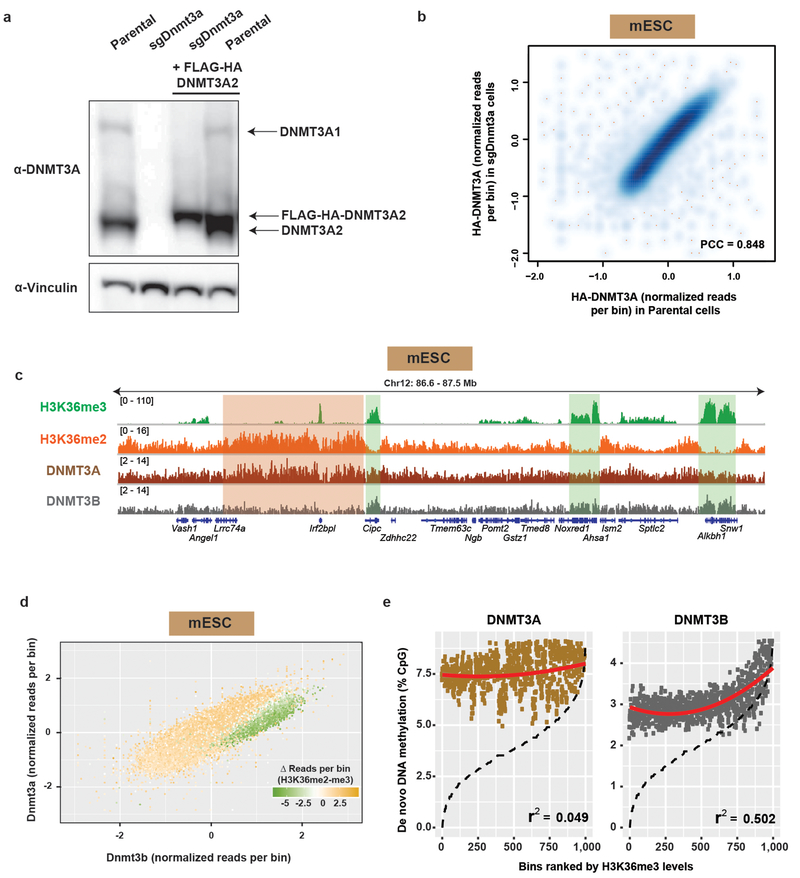
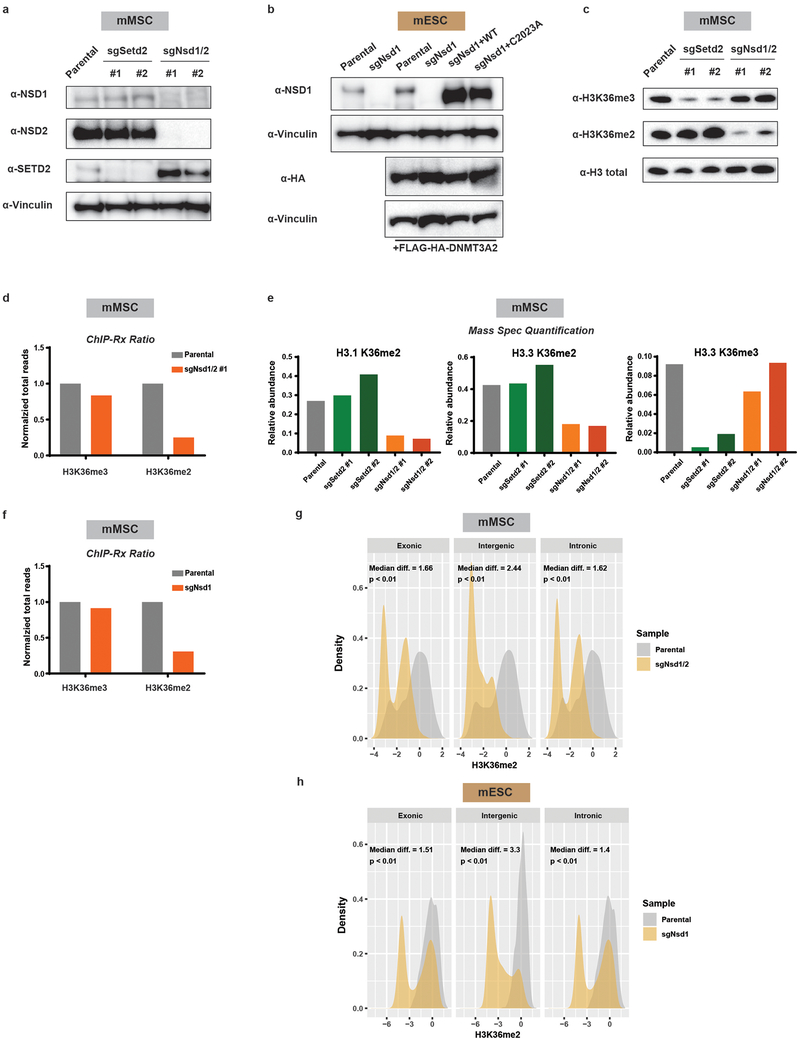
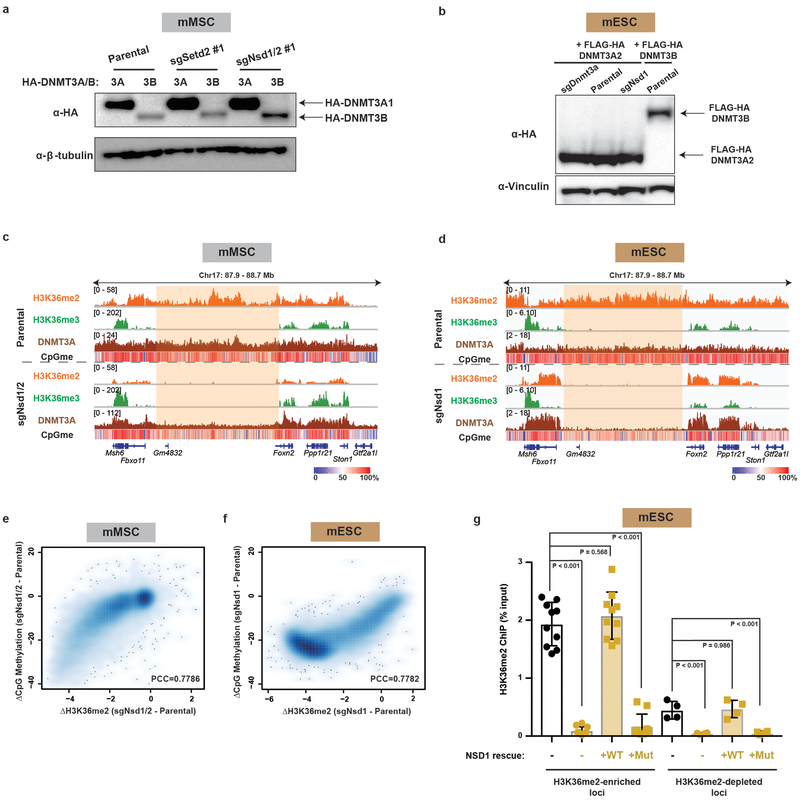
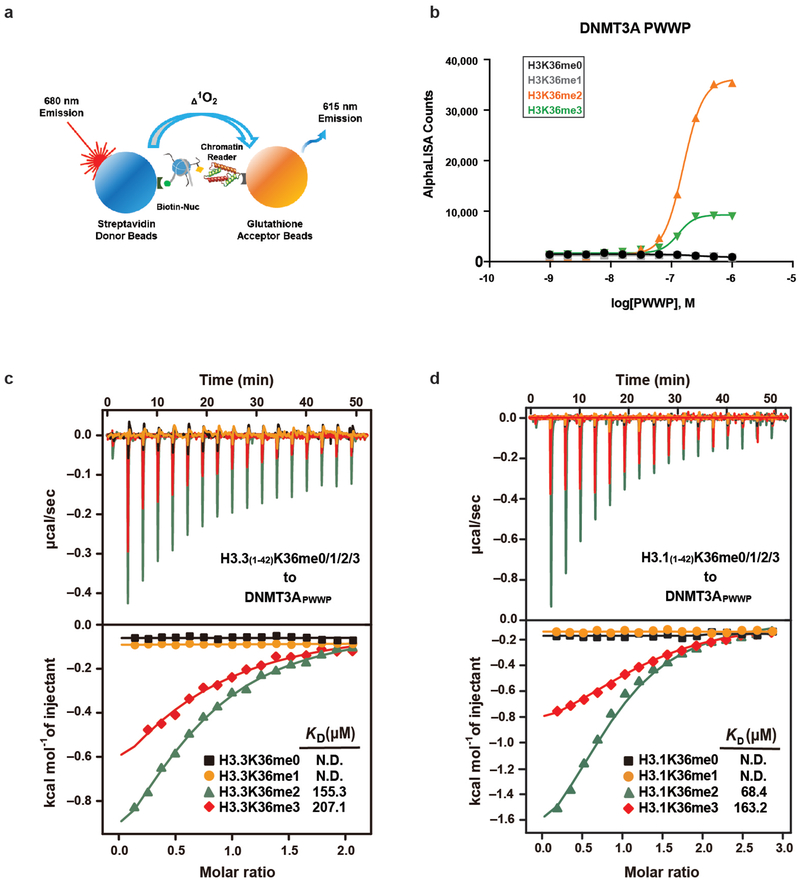
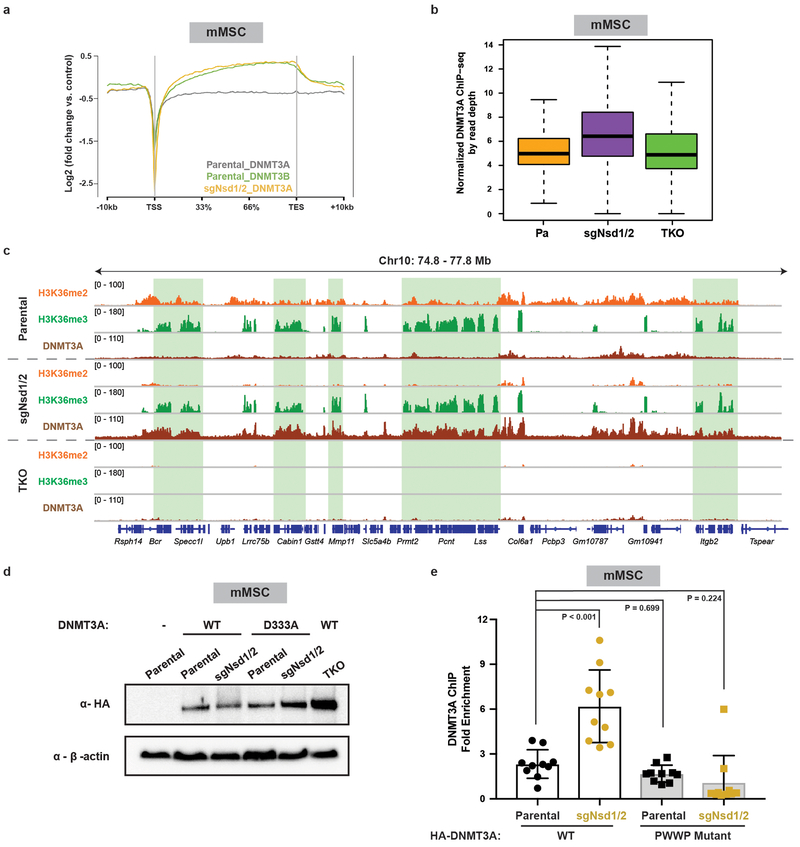
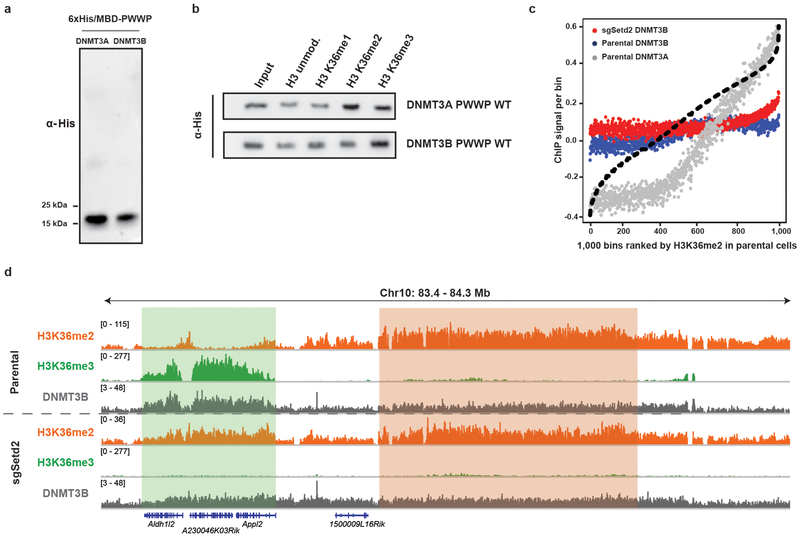
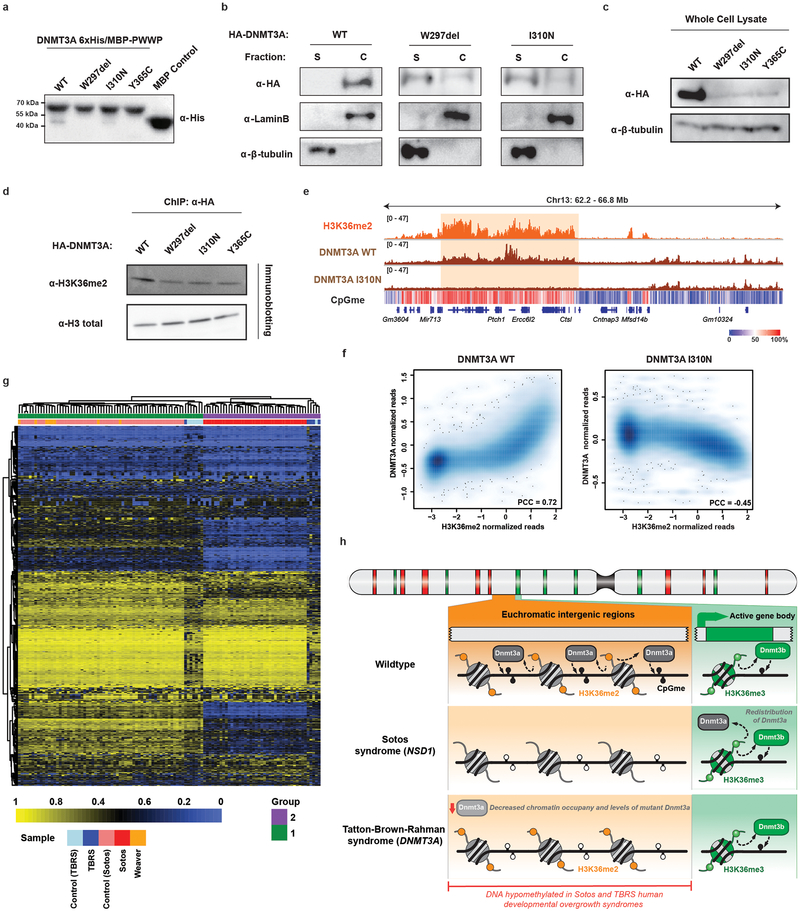
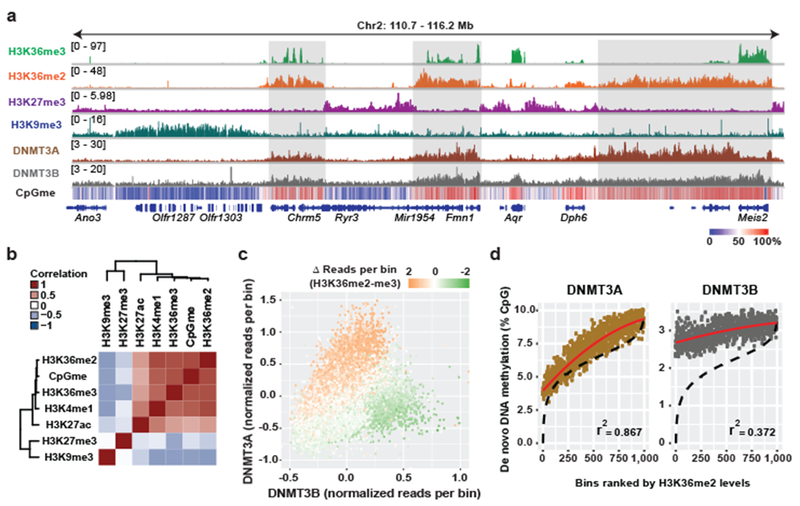
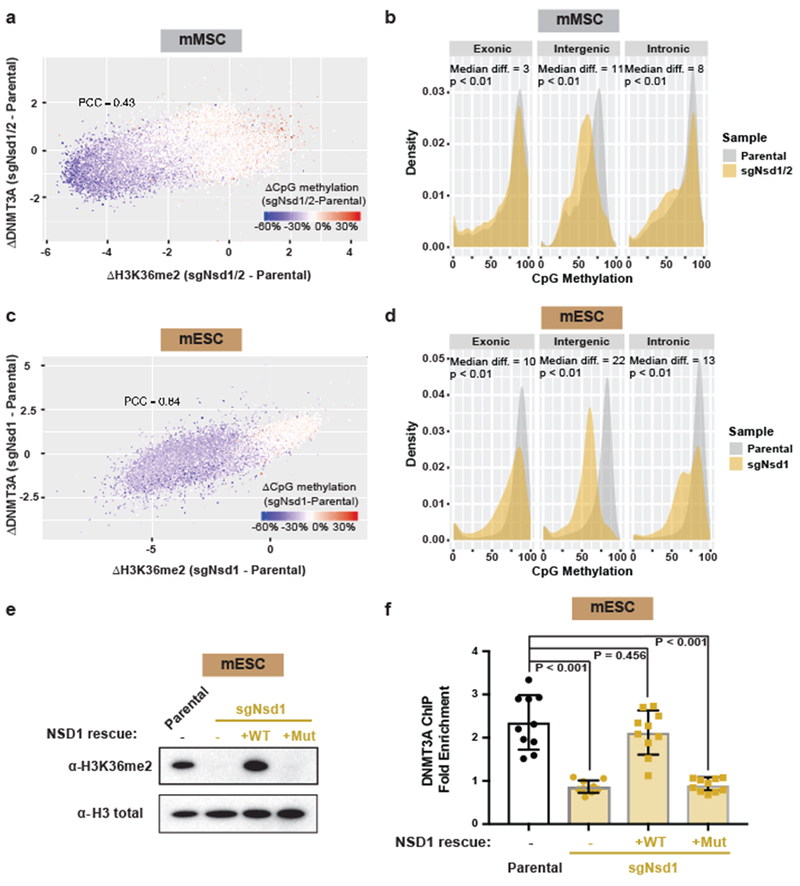
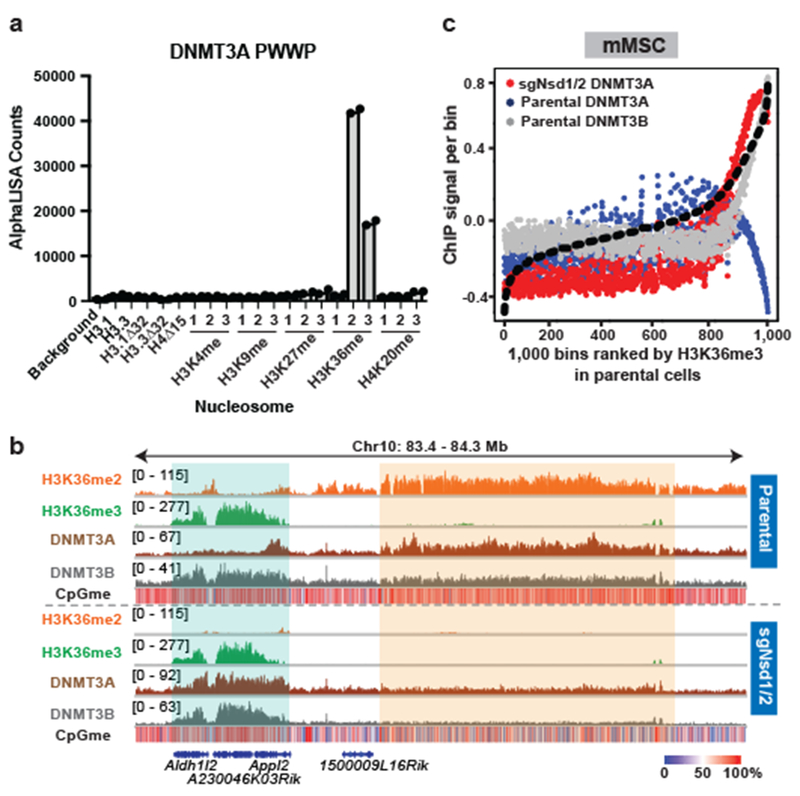
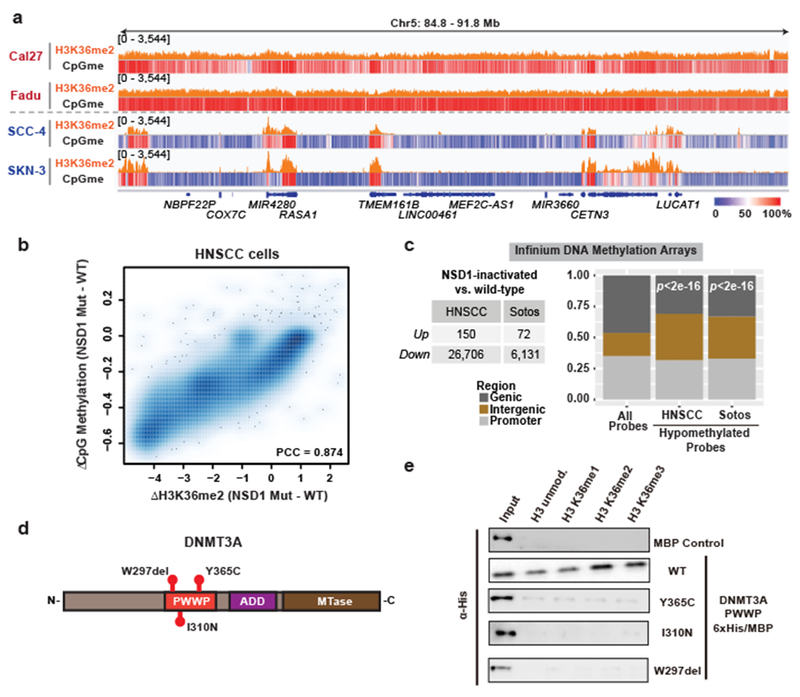
Enzymes that catalyse CpG methylation in DNA, including the DNA methyltransferases 1 (DNMT1), 3A (DNMT3A) and 3B (DNMT3B), are indispensable for mammalian tissue development and homeostasis1-4. They are also implicated in human developmental disorders and cancers5-8, supporting the critical role of DNA methylation in the specification and maintenance of cell fate. Previous studies have suggested that post-translational modifications of histones are involved in specifying patterns of DNA methyltransferase localization and DNA methylation at promoters and actively transcribed gene bodies9-11. However, the mechanisms that control the establishment and maintenance of intergenic DNA methylation remain poorly understood. Tatton-Brown-Rahman syndrome (TBRS) is a childhood overgrowth disorder that is defined by germline mutations in DNMT3A. TBRS shares clinical features with Sotos syndrome (which is caused by haploinsufficiency of NSD1, a histone methyltransferase that catalyses the dimethylation of histone H3 at K36 (H3K36me2)8,12,13), which suggests that there is a mechanistic link between these two diseases. Here we report that NSD1-mediated H3K36me2 is required for the recruitment of DNMT3A and maintenance of DNA methylation at intergenic regions. Genome-wide analysis shows that the binding and activity of DNMT3A colocalize with H3K36me2 at non-coding regions of euchromatin. Genetic ablation of Nsd1 and its paralogue Nsd2 in mouse cells results in a redistribution of DNMT3A to H3K36me3-modified gene bodies and a reduction in the methylation of intergenic DNA. Blood samples from patients with Sotos syndrome and NSD1-mutant tumours also exhibit hypomethylation of intergenic DNA. The PWWP domain of DNMT3A shows dual recognition of H3K36me2 and H3K36me3 in vitro, with a higher binding affinity towards H3K36me2 that is abrogated by TBRS-derived missense mutations. Together, our study reveals a trans-chromatin regulatory pathway that connects aberrant intergenic CpG methylation to human neoplastic and developmental overgrowth.
Conflict of interest statement
The authors declare competing financial interests. EpiCypher is a commercial developer and supplier of platforms similar to those used in this study: recombinant semi-synthetic modified nucleosomes and the dCypher nucleosome binding assay. H.C. and Y.Y. contributed equally as co-second authors to this manuscript.
Figures
References
-
- Li E, Bestor TH & Jaenisch R Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992). - PubMed
-
- Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). - PubMed
Methods References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
