

Molecular Strategies for RPGR Gene Therapy
- PMID: 31487940
- PMCID: PMC6770968
- DOI: 10.3390/genes10090674
Molecular Strategies for RPGR Gene Therapy
Abstract
Mutations affecting the Retinitis Pigmentosa GTPase Regulator (RPGR) gene are the commonest cause of X-linked and recessive retinitis pigmentosa (RP), accounting for 10%-20% of all cases of RP. The phenotype is one of the most severe amongst all causes of RP, characteristic for its early onset and rapid progression to blindness in young people. At present there is no cure for RPGR-related retinal disease. Recently, however, there have been important advances in RPGR research from bench to bedside that increased our understanding of RPGR function and led to the development of potential therapies, including the progress of adeno-associated viral (AAV)-mediated gene replacement therapy into clinical trials. This manuscript discusses the advances in molecular research, which have connected the RPGR protein with an important post-translational modification, known as glutamylation, that is essential for its optimal function as a key regulator of photoreceptor ciliary transport. In addition, we review key pre-clinical research that addressed challenges encountered during development of therapeutic vectors caused by high infidelity of the RPGR genomic sequence. Finally, we discuss the structure of three current phase I/II clinical trials based on three AAV vectors and RPGR sequences and link the rationale behind the use of the different vectors back to the bench research that led to their development.
Keywords: Retinitis Pigmentosa (RP); Retinitis Pigmentosa GTPase Regulator; adeno-associated viral; gene therapy.
Conflict of interest statement
REM receives grant funding from Nightstar Therapeutics (now Biogen Inc.). REM is a consultant to Nightstar Therapeutics and Spark Therapeutics. These companies did not have any input into the work presented. No other authors have a conflict of interest.
Figures

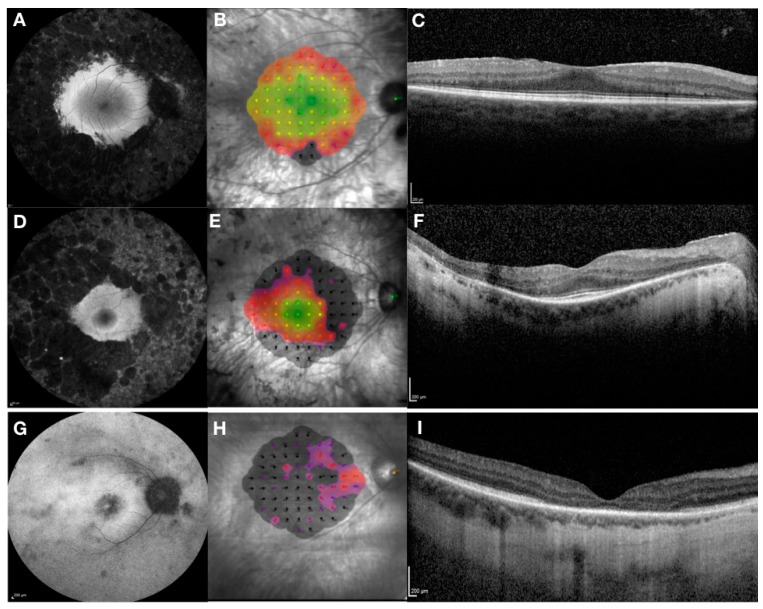


References
-
- Talib M., van Schooneveld M.J., Thiadens A.A., Fiocco M., Wijnholds J., Florijn R.J., Schalij-Delfos N.E., van Genderen M.M., Putter H., Cremers F.P.M., et al. Clinical and genetic characteristics of male patients with RPGR-associated retinal dystrophies: A long-term follow-up study. Retina. 2019;39:1186–1199. doi: 10.1097/IAE.0000000000002125. - DOI - PubMed
-
- Pelletier V., Jambou M., Delphin N., Zinovieva E., Stum M., Gigarel N., Dollfus H., Hamel C., Toutain A., Dufier J.L., et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: Genotype-phenotype correlations and impact on genetic counseling. Hum. Mutat. 2007;28:81–91. doi: 10.1002/humu.20417. - DOI - PubMed
-
- Branham K., Othman M., Brumm M., Karoukis A.J., Atmaca-Sonmez P., Yashar B.M., Schwartz S.B., Stover N.B., Trzupek K., Wheaton D., et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Investig. Ophthalmol. Vis. Sci. 2012;53:8232–8237. doi: 10.1167/iovs.12-11025. - DOI - PMC - PubMed
-
- Webb T.R., Parfitt D.A., Gardner J.C., Martinez A., Bevilacqua D., Davidson A.E., Zito I., Thiselton D.L., Ressa J.H., Apergi M., et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum. Mol. Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
