

Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues
- PMID: 31487953
- PMCID: PMC6780404
- DOI: 10.3390/jcm8091385
Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues
Abstract
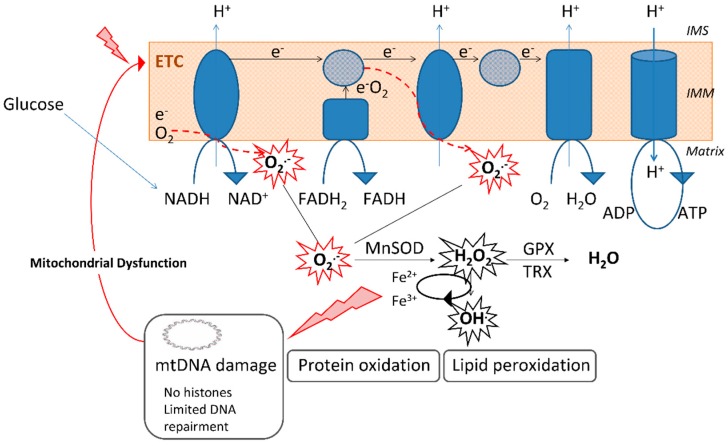
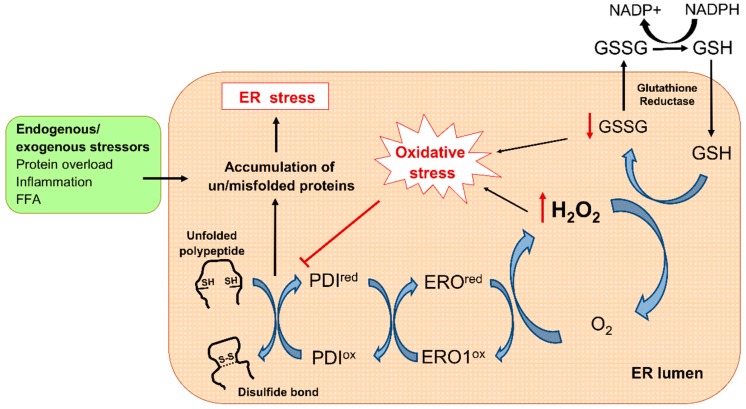
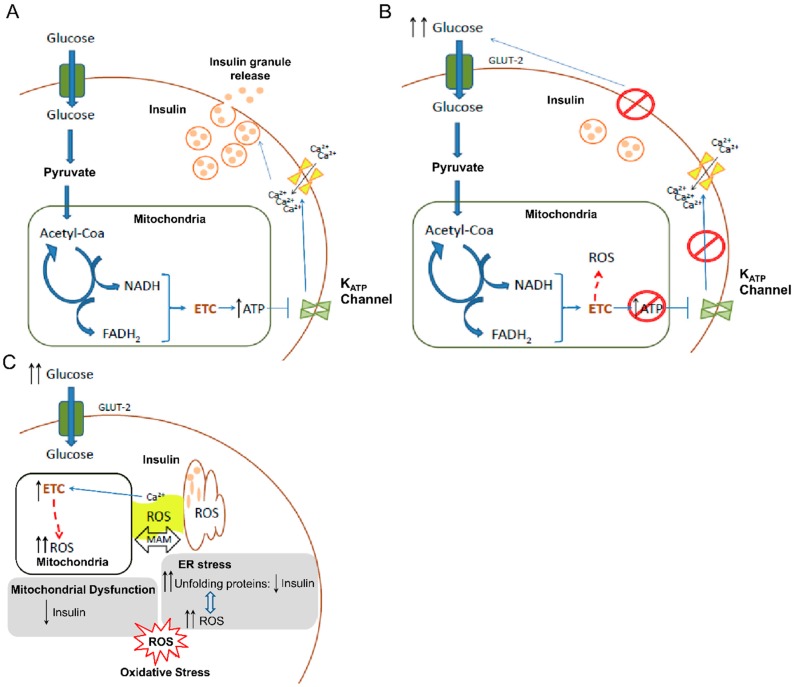
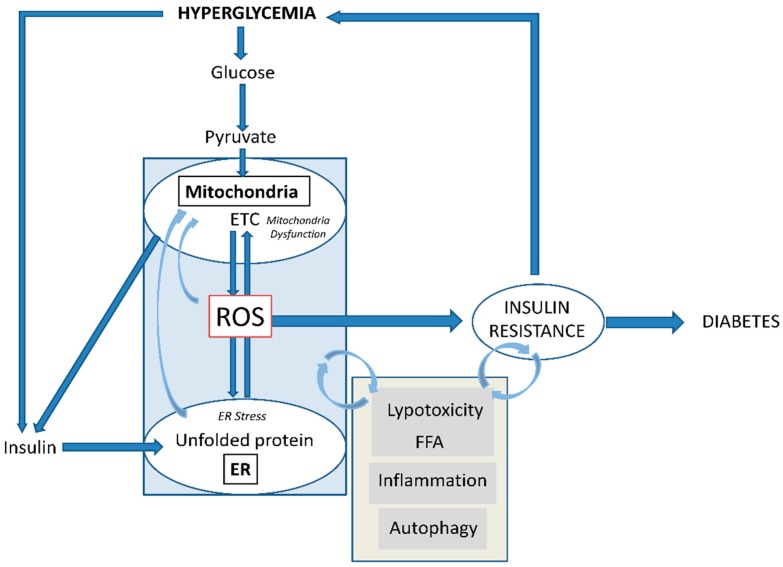
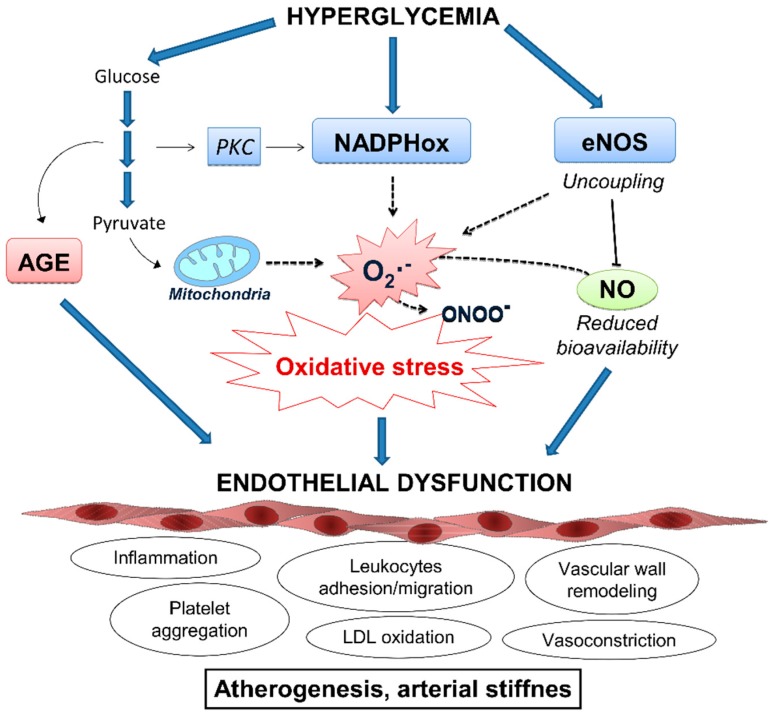
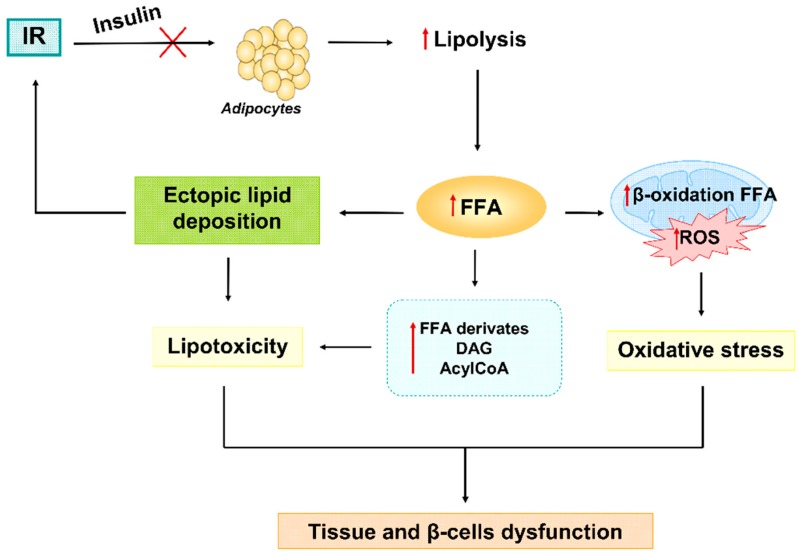
Type 2 diabetes (T2D) is a metabolic disorder characterized by hyperglycemia and insulin resistance in which oxidative stress is thought to be a primary cause. Considering that mitochondria are the main source of ROS, we have set out to provide a general overview on how oxidative stress is generated and related to T2D. Enhanced generation of reactive oxygen species (ROS) and oxidative stress occurs in mitochondria as a consequence of an overload of glucose and oxidative phosphorylation. Endoplasmic reticulum (ER) stress plays an important role in oxidative stress, as it is also a source of ROS. The tight interconnection between both organelles through mitochondrial-associated membranes (MAMs) means that the ROS generated in mitochondria promote ER stress. Therefore, a state of stress and mitochondrial dysfunction are consequences of this vicious cycle. The implication of mitochondria in insulin release and the exposure of pancreatic β-cells to hyperglycemia make them especially susceptible to oxidative stress and mitochondrial dysfunction. In fact, crosstalk between both mechanisms is related with alterations in glucose homeostasis and can lead to the diabetes-associated insulin-resistance status. In the present review, we discuss the current knowledge of the relationship between oxidative stress, mitochondria, ER stress, inflammation, and lipotoxicity in T2D.
Keywords: ER stress; ROS; antioxidants; insulin resistance; mitochondria; oxidative stress; type 2 diabetes.
Conflict of interest statement
References
Figures
References
Publication types
Grants and funding
- PI16/1083, PI16/0301, FI17/00144, FI17/00126, CIBERehd CB06/04/0071, CPII16/00037/Instituto de Salud Carlos III
- 'A way to build Europe'/European Regional Development Fund (ERDF)
- UGP15-193, UGP-15-220, UGP-15-144/Foundation for the Promotion of Health and Biomedical Research of Valencia Region
- GRISOLIAP/2016/015/Valencian Regional Government
- CES10/030/Ministry of Health of the Valencian Regional Government
LinkOut - more resources
Full Text Sources
Medical
