

Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data
- PMID: 31488068
- PMCID: PMC6727555
- DOI: 10.1186/s12859-019-3031-y
Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data
Abstract
Background: Metagenomics caused a quantum leap in microbial ecology. However, the inherent size and complexity of metagenomic data limit its interpretation. The quantification of metagenomic traits in metagenomic analysis workflows has the potential to improve the exploitation of metagenomic data. Metagenomic traits are organisms' characteristics linked to their performance. They are measured at the genomic level taking a random sample of individuals in a community. As such, these traits provide valuable information to uncover microorganisms' ecological patterns. The Average Genome Size (AGS) and the 16S rRNA gene Average Copy Number (ACN) are two highly informative metagenomic traits that reflect microorganisms' ecological strategies as well as the environmental conditions they inhabit.
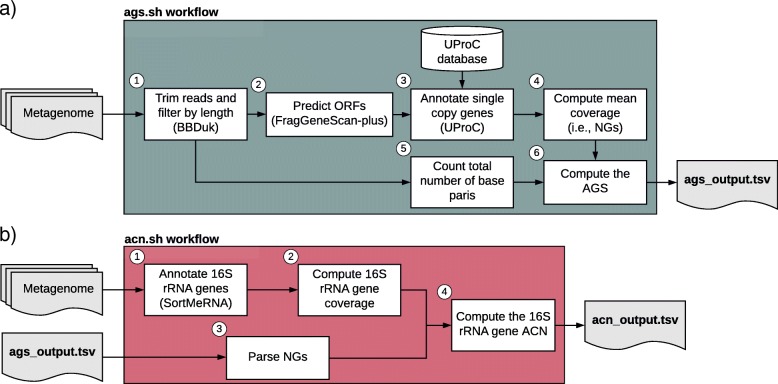
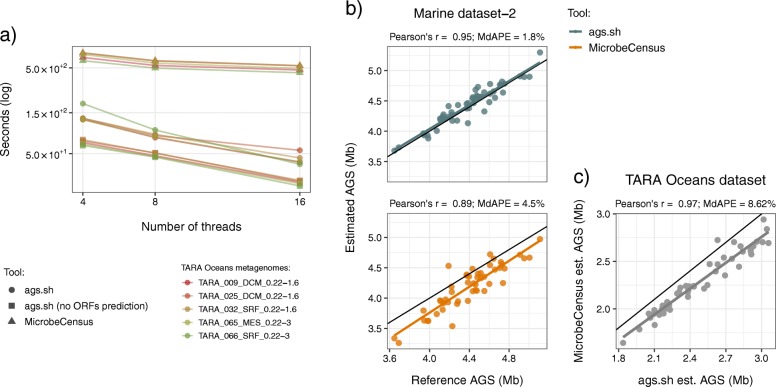
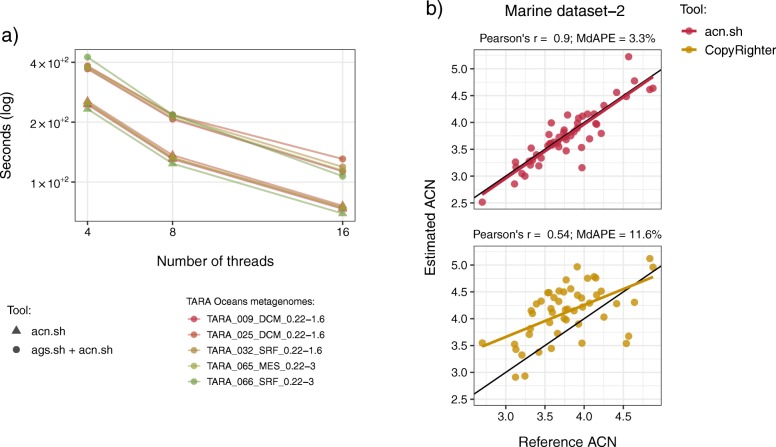
Results: Here, we present the ags.sh and acn.sh tools, which analytically derive the AGS and ACN metagenomic traits. These tools represent an advance on previous approaches to compute the AGS and ACN traits. Benchmarking shows that ags.sh is up to 11 times faster than state-of-the-art tools dedicated to the estimation AGS. Both ags.sh and acn.sh show comparable or higher accuracy than existing tools used to estimate these traits. To exemplify the applicability of both tools, we analyzed the 139 prokaryotic metagenomes of TARA Oceans and revealed the ecological strategies associated with different water layers.
Conclusion: We took advantage of recent advances in gene annotation to develop the ags.sh and acn.sh tools to combine easy tool usage with fast and accurate performance. Our tools compute the AGS and ACN metagenomic traits on unassembled metagenomes and allow researchers to improve their metagenomic data analysis to gain deeper insights into microorganisms' ecology. The ags.sh and acn.sh tools are publicly available using Docker container technology at https://github.com/pereiramemo/AGS-and-ACN-tools .
Keywords: 16S rRNA gene average copy number; Average genome size; Functional traits; Metagenomics; Microbial ecology.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
