

Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice
- PMID: 31489936
- PMCID: PMC6770546
- DOI: 10.3390/nu11092127
Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice
Abstract
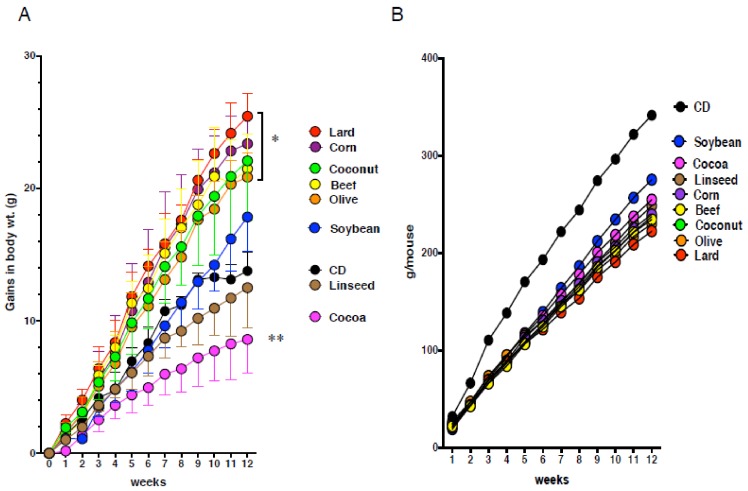
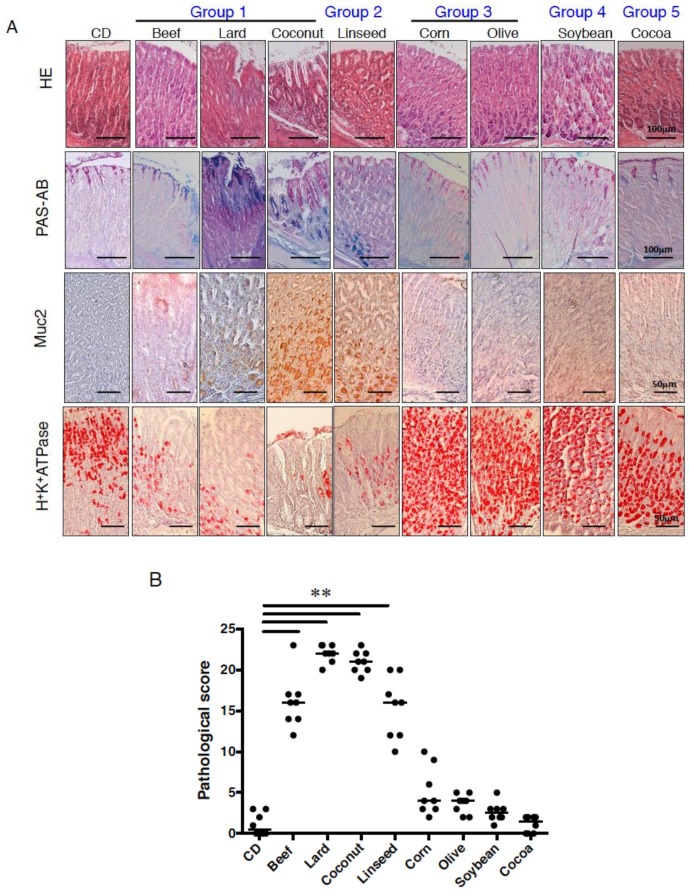
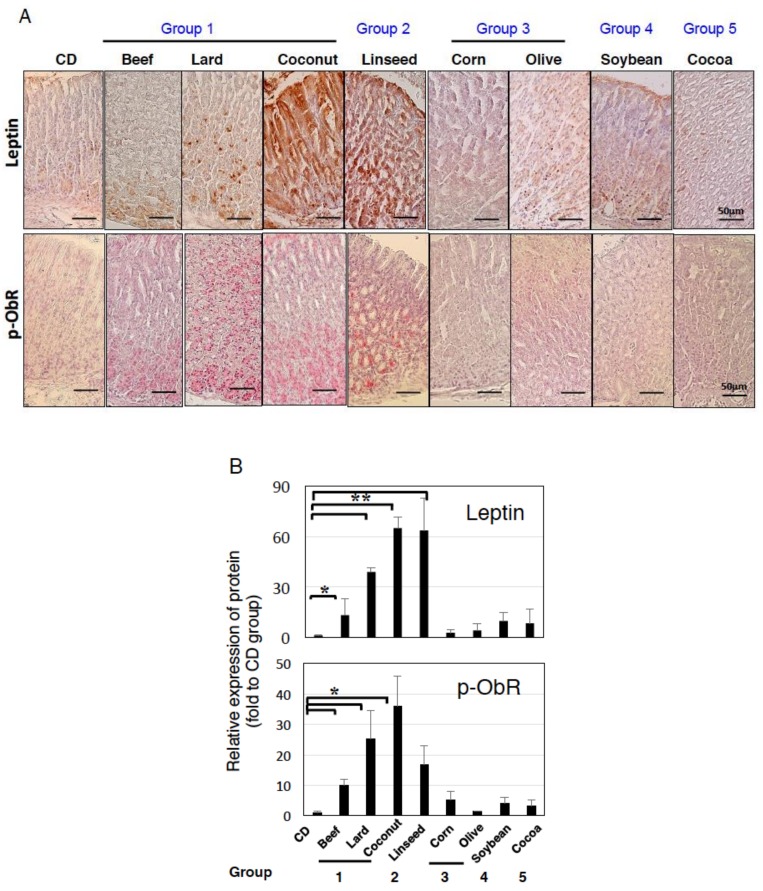
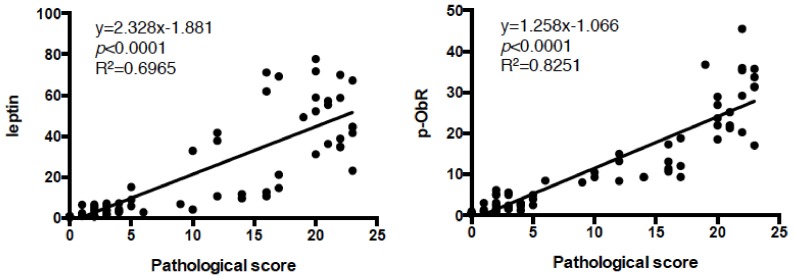
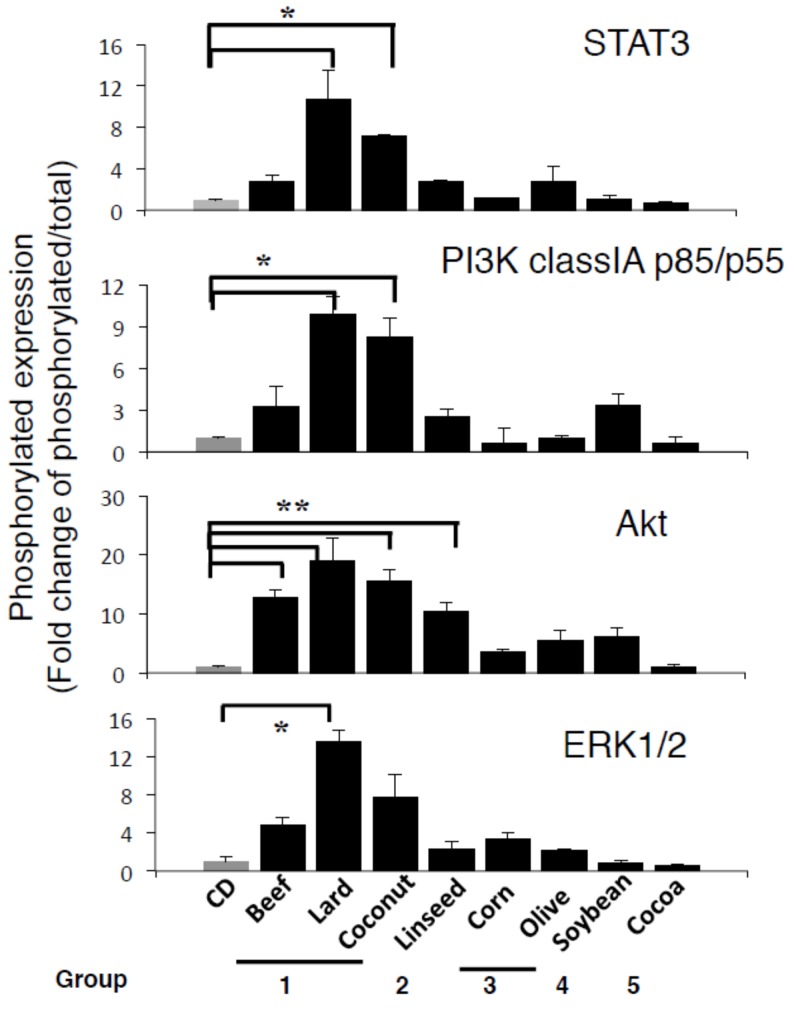
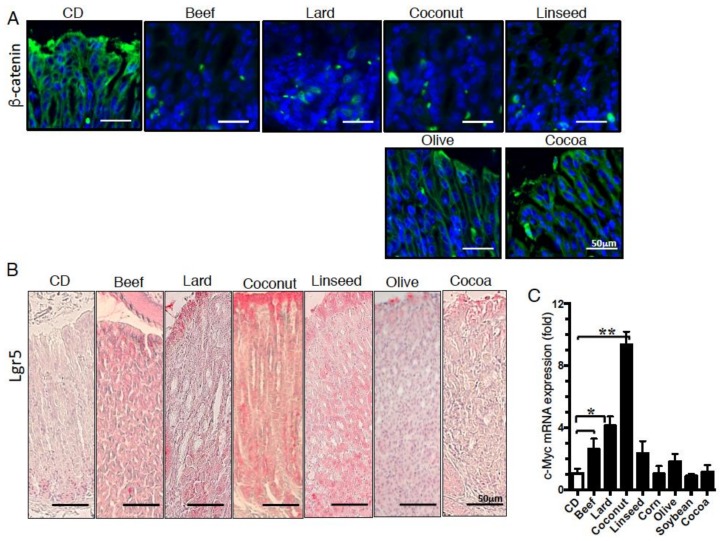
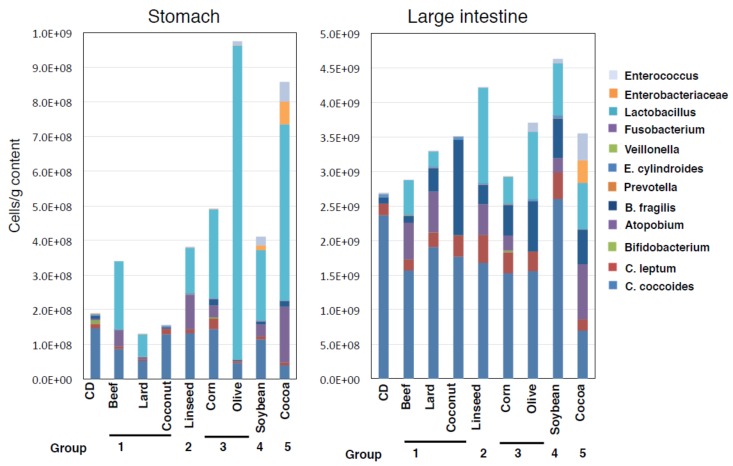
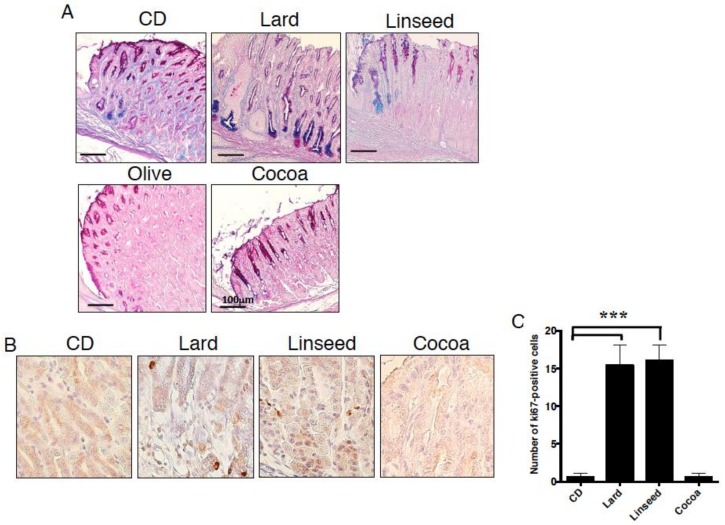
Excess of fat intake leads to obesity and causes a variety of metabolic diseases and cancer. We previously demonstrated that high-lard diet induces intestinal metaplasia, a precancerous lesion of the stomach mediated by leptin signaling. This study aims to investigate which kinds of dietary fat cause the intestinal metaplasia onset. We fed eight kinds of high-fat diets (HFDs) of animal or plant origin to mice evaluated their effect on gastric pathogenesis. Five types of dietary fat were divided according to their observed effects: Obese with high metaplasia (group I; beef tallow, lard, and hydrogenated coconut oil), non-obese with high metaplasia (group II; linseed oil), obese without metaplasia (group III; corn oil and olive oil), non-obese without metaplasia (group IV, soybean oil) and lean without metaplasia (group V; cocoa butter). The group I and II diets induced leptin, phosphorylated leptin receptor (ObR), signal transducer and activator 3 (STAT3), and increased intracellular β-catenin accumulation in the stomach. Moreover, mice fed these HFDs with 1-methyl-3-nitro-1-nitrosoguanidine (MNNG), a gastric carcinogen, and further accelerated dysplasia in the stomach. Lactobacillus occupancy in the stomach increased in all HFDs except hydrogenated coconut oil. Our findings suggest that HFDs inducing leptin signaling accelerate the enhancement of protumorigenic gastric microenvironment independent of body mass gain or microbiome changes.
Keywords: high-fat; leptin; microbiota; protumorigenesis; stomach.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
