

doi: 10.1016/j.ajhg.2019.07.011.
Mendelian Gene Discovery: Fast and Furious with No End in Sight
Affiliations
- PMID: 31491408
- PMCID: PMC6731362
- DOI: 10.1016/j.ajhg.2019.07.011
Item in Clipboard
Mendelian Gene Discovery: Fast and Furious with No End in Sight
Am J Hum Genet.
.
Abstract
Gene discovery for Mendelian conditions (MCs) offers a direct path to understanding genome function. Approaches based on next-generation sequencing applied at scale have dramatically accelerated gene discovery and transformed genetic medicine. Finding the genetic basis of ∼6,000-13,000 MCs yet to be delineated will require both technical and computational innovation, but will rely to a larger extent on meaningful data sharing.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
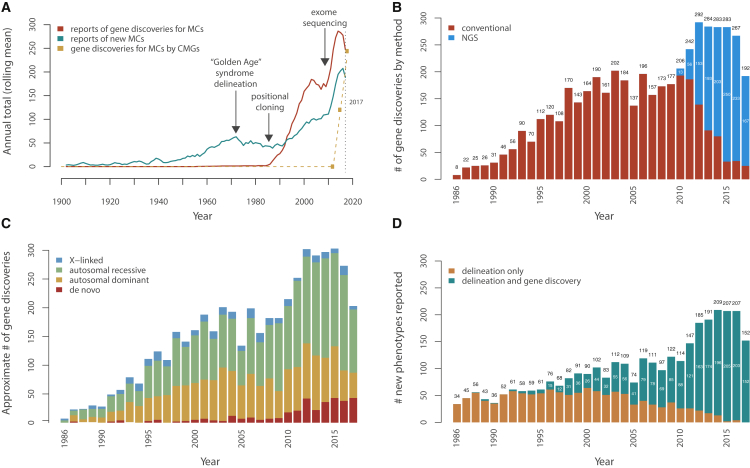
Annualized Metrics of Gene Discovery for Mendelian Conditions (A) Approximate rates of reported gene discoveries for Mendelian conditions (MCs) and of delineation of MCs over time (1900–2017). Trends in reported (i.e., published) delineations of new MCs, including the so-called “Golden Age” of syndrome delineation in the 1970s, leading to a peak throughout that decade. These data also show the impact of technical and methodological advances that fueled gene discovery, namely the impact of positional cloning in 1986; the development of dense, genome-wide linkage maps in the early 1990s; and increasing knowledge, gained via the Human Genome Project (1990–2001), of the physical locations and sequence content of genes. Rates shown for gene discoveries and syndrome delineations reflect publications, not unpublished discoveries or syndrome delineations, as recorded in OMIM and extracted by text analysis. The dashed line represents the number of genes for MCs reported discovered by the Centers of Mendelian Genomics, most of which are unpublished. (B) Approximate number of gene discoveries per year for MCs made by exome sequencing (ES) and next-generation sequencing (NGS) versus conventional approaches. Following the introduction of positional cloning in 1986 and of ES in 2010, there were rapid increases in the rate of gene discovery for MCs. Each new approach made gene discovery possible for MCs that had otherwise been intractable to prior approaches, and this added to the baseline rate of gene discovery. Since 2010, NGS-based approaches (blue) have been used to make nearly all gene discoveries for MCs compared to conventional approaches (red). (C) Approximate number of gene discoveries per year for MCs by mode of inheritance. The estimated proportion of gene discoveries for MCs due to de novo variants (DNVs, red) has increased since 2010 as NGS made routine identification of such variants possible (see Supplemental Methods in Supplemental Data). However, the proportions of gene discoveries for autosomal recessive (green), dominant (orange), and X-linked (blue) MCs each continue to be equal to or greater than the number of discoveries for MCs due to de novo (red) variants. Moreover, until 2010, the vast majority of gene discoveries for MCs were for inherited conditions (∼97% before 2010; ∼89% from 2010–2016; and ∼79% in 2017), so still, most MCs known to date (∼90%–93%) are predominately due to inherited variants. MCs assessed as being attributable to DNVs were excluded from the autosomal dominant and X-linked groups and vice versa, and MCs attributed to both dominant and recessive variants are not shown. Modes of inheritance were inferred by text analysis of OMIM entries. (D) Impact of ES and NGS on the rate and method of syndrome delineation. Classical syndrome delineation (orange) is phenotype-driven and proceeds by ascertaining multiple individuals with overlapping clinical findings and then identifying of the underlying gene. In contrast, for genotype-driven syndrome delineation (teal), persons with overlapping clinical findings are identified only after discovery that they share pathogenic variants in the same candidate gene. Introduction of NGS-based approaches rapidly extinguished phenotype-driven syndrome delineation, and as of 2017, new MCs have been reported only after discovery of the underlying gene. MCs for which the gene was discovered in the same year as the first publication of data from an individual with the MC were categorized as genotype-driven; MCs for which data on the first individual with the MC was published one year or more prior to gene discovery were categorized as phenotype-driven (see Supplemental Methods in Supplemental Data).
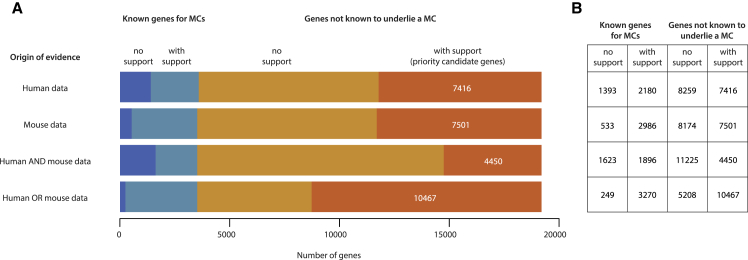
Estimated Number of Genes for Mendelian Conditions (A) Priority candidate genes for Mendelian conditions (MCs) yet to be discovered or delineated can be identified based on detecting a deficit of variation in healthy human controls due to purifying selection (i.e., human data) or the existence of at least one mutant mouse line with an abnormal phenotype (i.e., mouse data). Each stacked bar illustrates, from left to right, the number of human genes known to underlie an MC but not supported by a given type of evidence (dark blue); known to underlie an MC and supported by evidence (light blue); not known to underlie an MC (i.e., novel) and not supported by evidence (burnt orange); and not known to underlie an MC but evidence suggests that it does and it is therefore a priority candidate gene (bright orange) supported by evidence. Selecting the intersection of genes supported by both mouse and human data yields 4,450 priority candidate genes that are likely to underlie one or more novel MCs. (B) Alternatively, the union of genes supported by either mouse or human data suggests there are at least 10,467 priority candidate genes likely to underlie one or more new MCs. Both of these estimates are probably conservative for several reasons: ∼25% of genes underlie two or more distinct MCs (adjusting for this yields ∼6,100–14,400 potential novel MCs), mutant phenotypes for >12,000 mouse genes have not yet been assessed, and current human constraint metrics still lack power to detect constraint in ∼30% of all genes and are underpowered to detect constraint against homozygous loss of function. (See Supplemental Data for details.)
References
-
- Antonarakis S.E., Beckmann J.S. Mendelian disorders deserve more attention. Nat. Rev. Genet. 2006;7:277–282. - PubMed
-
- Starita L.M., Islam M.M., Banerjee T., Adamovich A.I., Gullingsrud J., Fields S., Shendure J., Parvin J.D. A multiplex homology-directed DNA repair assay reveals the impact of more than 1,000 BRCA1 missense substitution variants on protein function. Am. J. Hum. Genet. 2018;103:498–508. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
