

Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study
- PMID: 31491988
- PMCID: PMC6770978
- DOI: 10.3390/genes10090678
Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study
Abstract
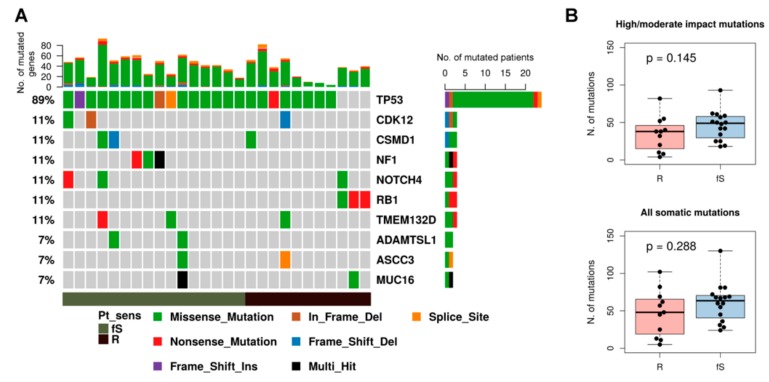
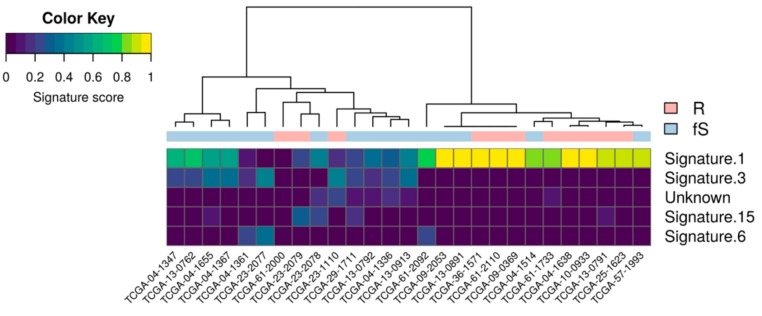
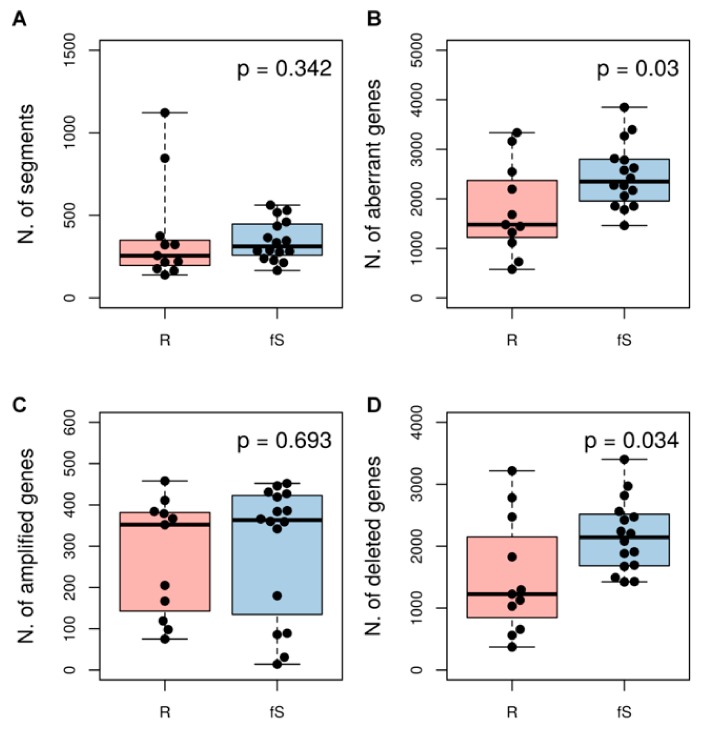
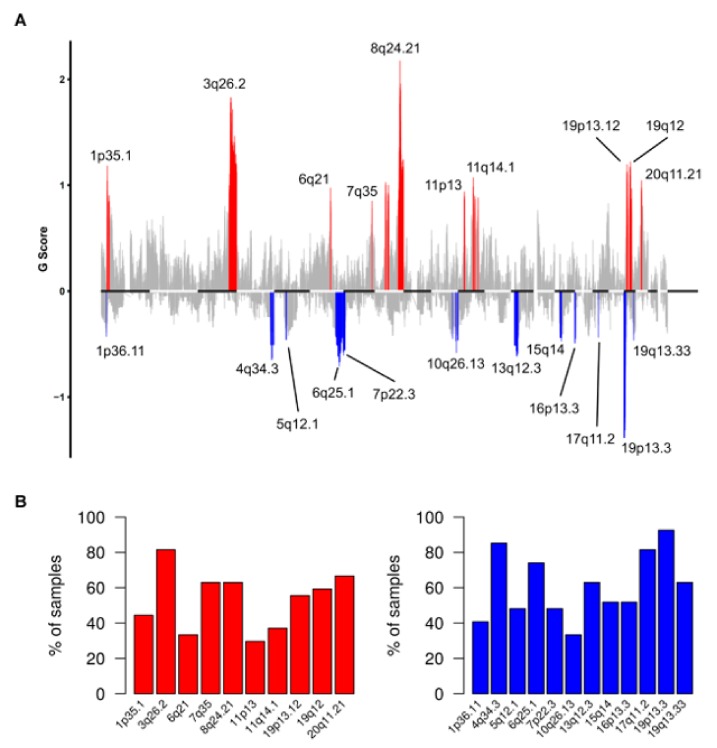
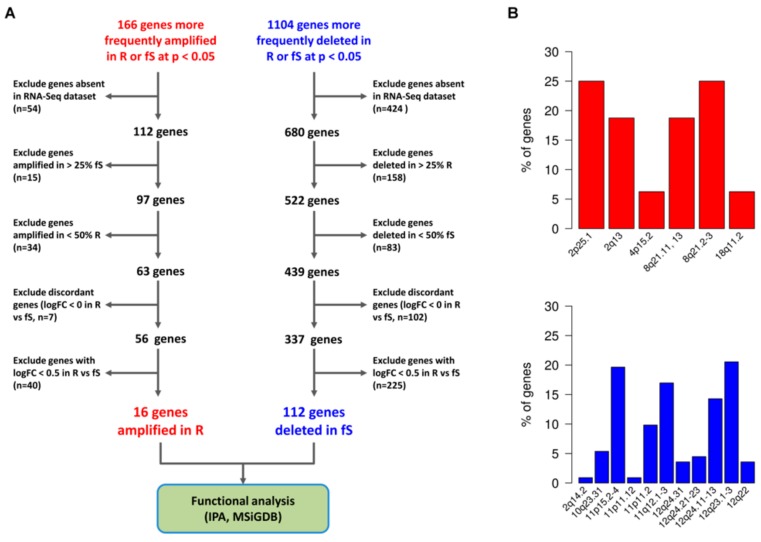
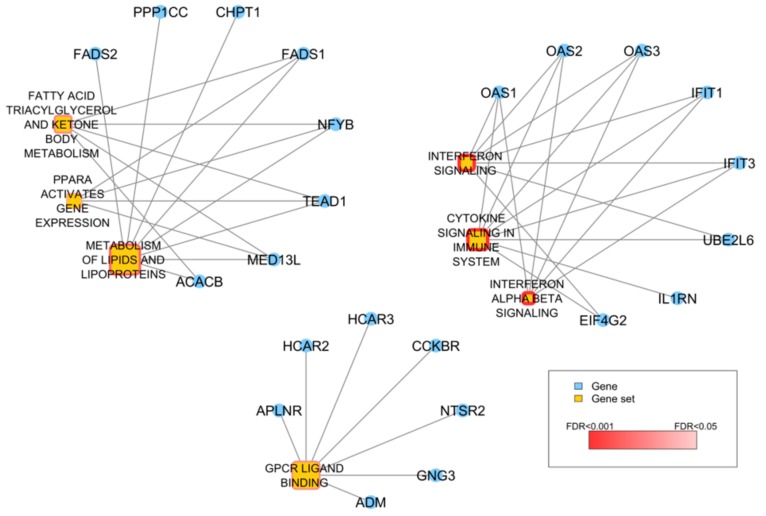
High grade serous ovarian cancer (HGSOC) retains high molecular heterogeneity and genomic instability, which currently limit the treatment opportunities. HGSOC patients receiving complete cytoreduction (R0) at primary surgery and platinum-based therapy may unevenly experience early disease relapse, in spite of their clinically favorable prognosis. To identify distinctive traits of the genomic landscape guiding tumor progression, we focused on the R0 patients of The Cancer Genome Atlas (TCGA) ovarian serous cystadenocarcinoma (TCGA-OV) dataset and classified them according to their time to relapse (TTR) from surgery. We included in the study two groups of R0-TCGA patients experiencing substantially different outcome: Resistant (R; TTR ≤ 12 months; n = 11) and frankly Sensitive (fS; TTR ≥ 24 months; n = 16). We performed an integrated clinical, RNA-Sequencing, exome and somatic copy number alteration (sCNA) data analysis. No significant differences in mutational landscape were detected, although the lack of BRCA-related mutational signature characterized the R group. Focal sCNA analysis showed a higher frequency of amplification in R group and deletions in fS group respectively, involving cytobands not commonly detected by recurrent sCNA analysis. Functional analysis of focal sCNA with a concordantly altered gene expression identified in R group a gain in Notch, and interferon signaling and fatty acid metabolism. We are aware of the constraints related to the low number of OC cases analyzed. It is worth noting, however, that the sCNA identified in this exploratory analysis and characterizing Pt-resistance are novel, deserving validation in a wider cohort of patients achieving complete surgical debulking.
Keywords: focal copy number alterations; ovarian cancer; platinum resistance; whole exome sequencing.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
