

Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia
- PMID: 31492675
- PMCID: PMC6888143
- DOI: 10.1182/blood.2019000015
Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia
Abstract
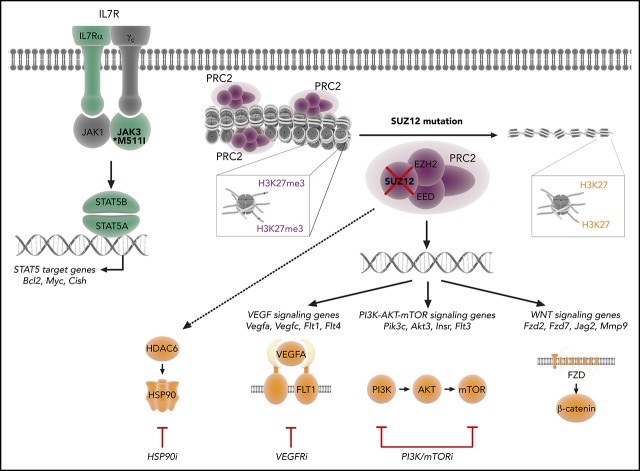
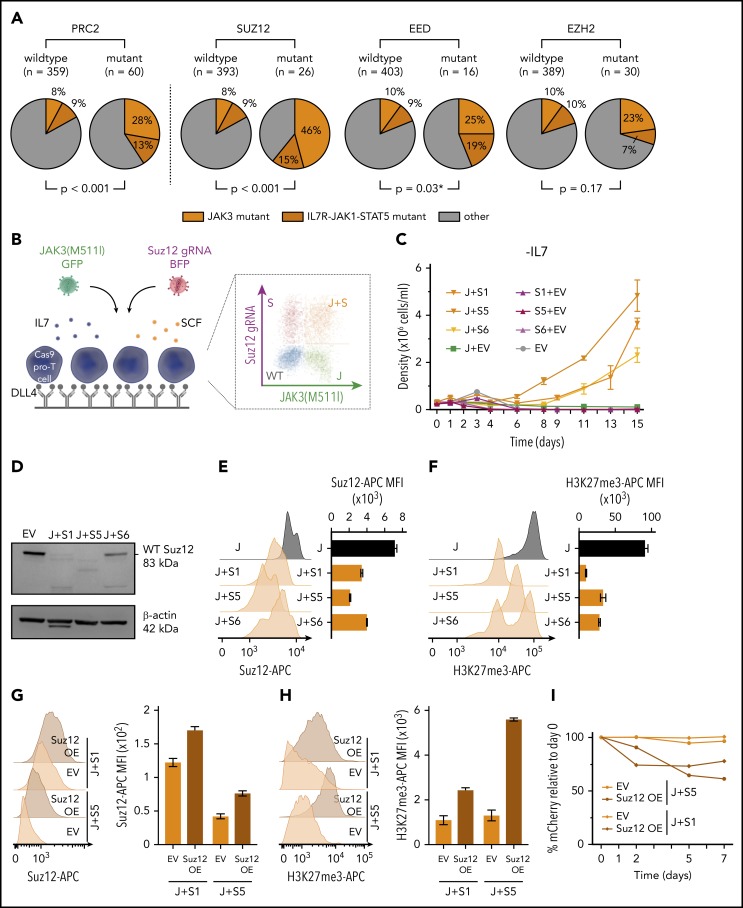
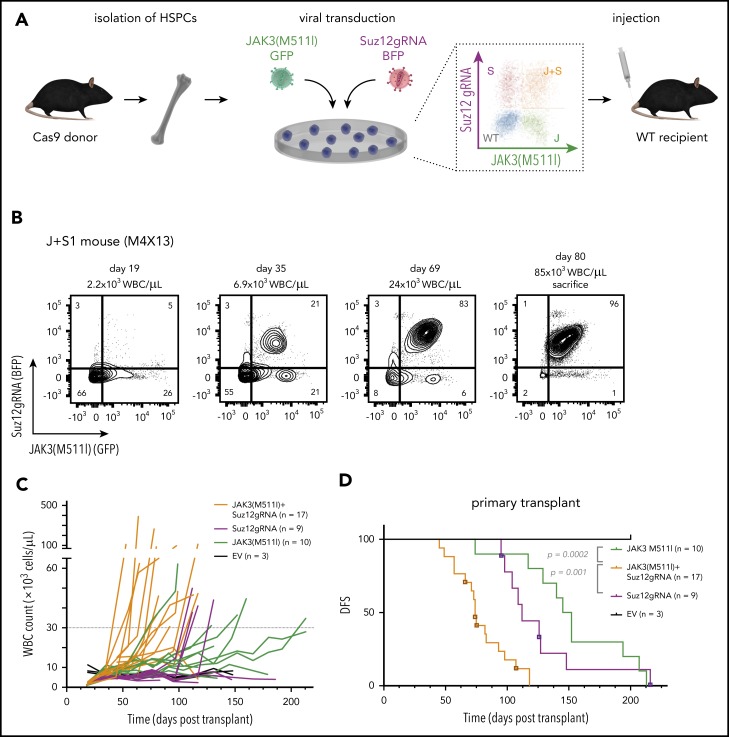
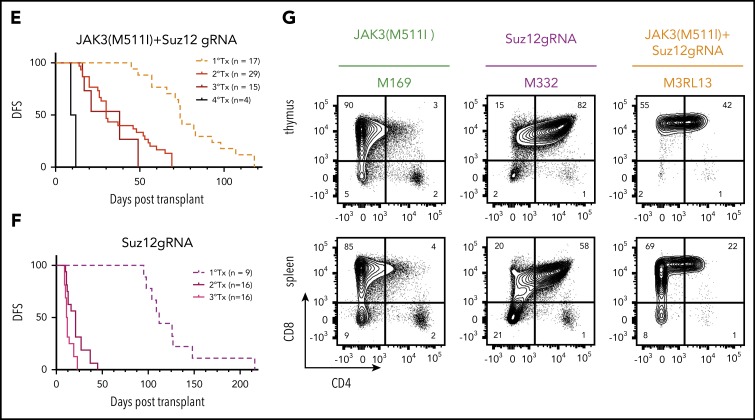
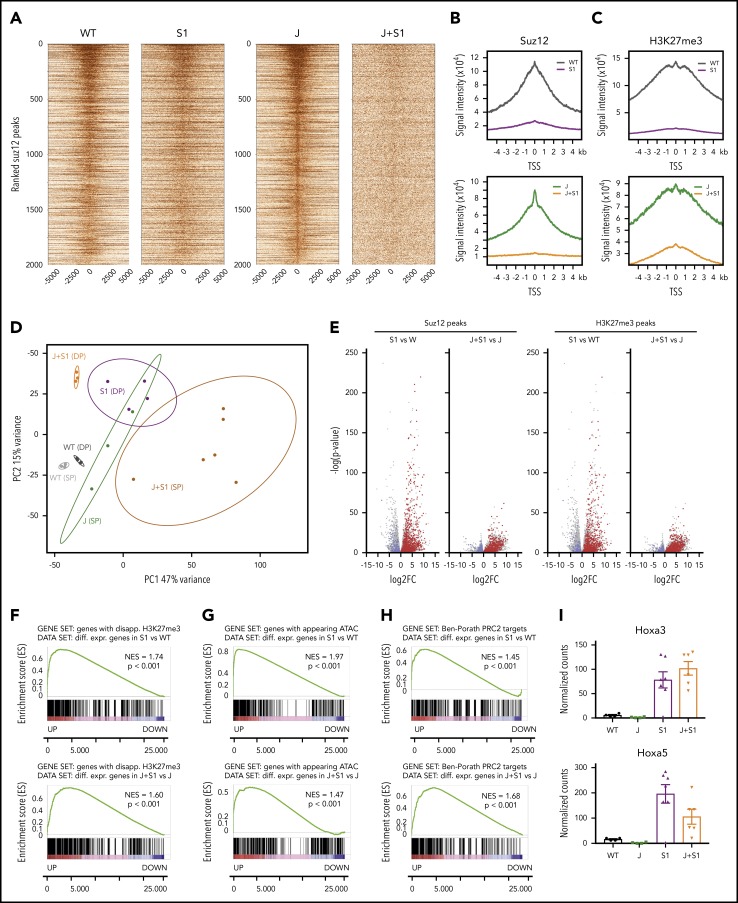
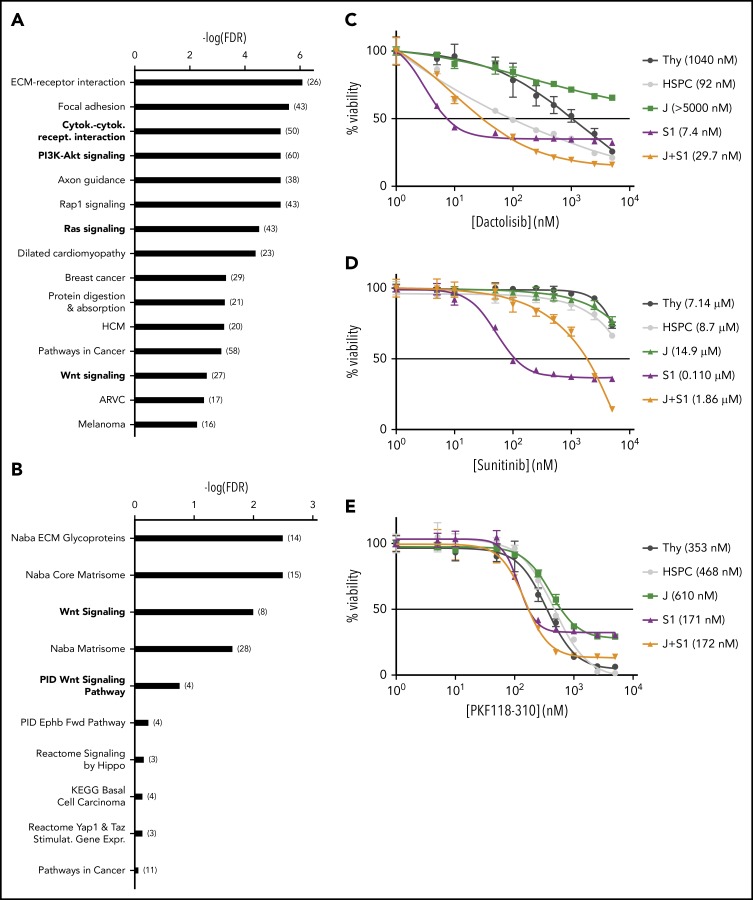
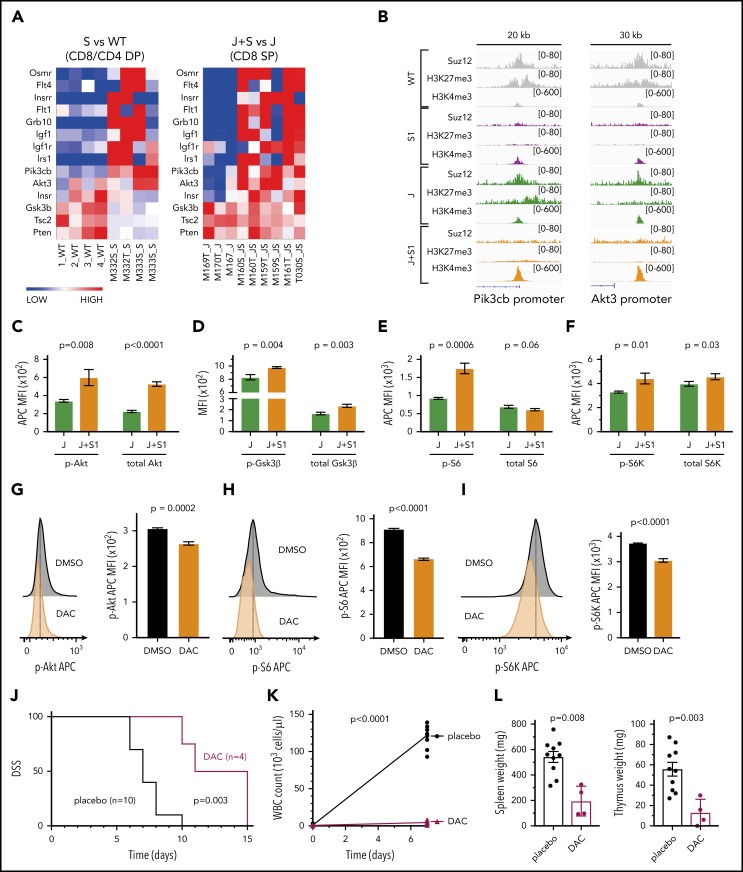
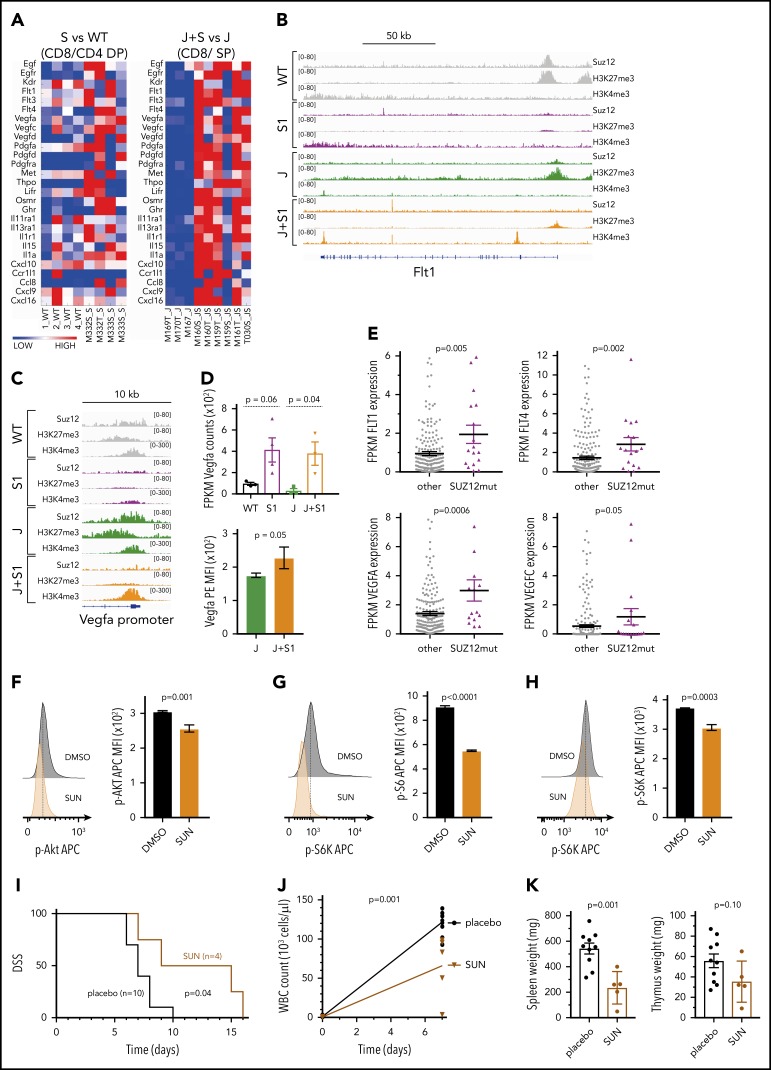
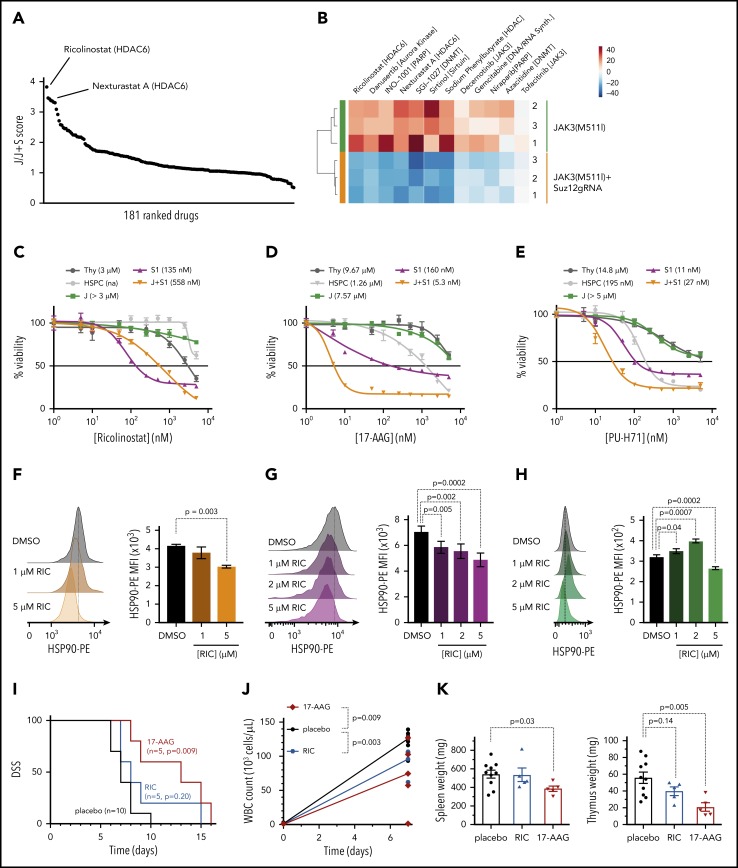
The polycomb repressive complex 2, with core components EZH2, SUZ12, and EED, is responsible for writing histone 3 lysine 27 trimethylation histone marks associated with gene repression. Analysis of sequence data from 419 T-cell acute lymphoblastic leukemia (T-ALL) cases demonstrated a significant association between SUZ12 and JAK3 mutations. Here we show that CRISPR/Cas9-mediated inactivation of Suz12 cooperates with mutant JAK3 to drive T-cell transformation and T-ALL development. Gene expression profiling integrated with ChIP-seq and ATAC-seq data established that inactivation of Suz12 led to increased PI3K/mammalian target of rapamycin (mTOR), vascular endothelial growth factor (VEGF), and WNT signaling. Moreover, a drug screen revealed that JAK3/Suz12 mutant leukemia cells were more sensitive to histone deacetylase (HDAC)6 inhibition than JAK3 mutant leukemia cells. Among the broad genome and gene expression changes observed on Suz12 inactivation, our integrated analysis identified the PI3K/mTOR, VEGF/VEGF receptor, and HDAC6/HSP90 pathways as specific vulnerabilities in T-ALL cells with combined JAK3 and SUZ12 mutations.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16(12):803-810. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
