

Preclinical Testing of Nalfurafine as an Opioid-sparing Adjuvant that Potentiates Analgesia by the Mu Opioid Receptor-targeting Agonist Morphine
- PMID: 31492823
- PMCID: PMC6863463
- DOI: 10.1124/jpet.118.255661
Preclinical Testing of Nalfurafine as an Opioid-sparing Adjuvant that Potentiates Analgesia by the Mu Opioid Receptor-targeting Agonist Morphine
Abstract
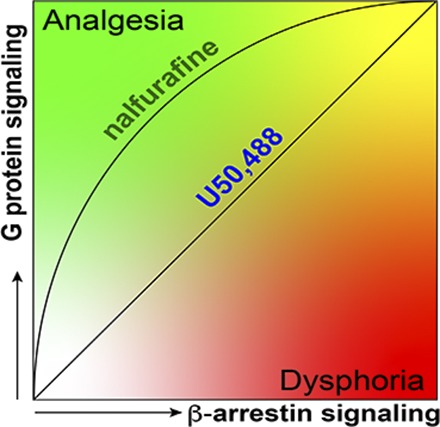
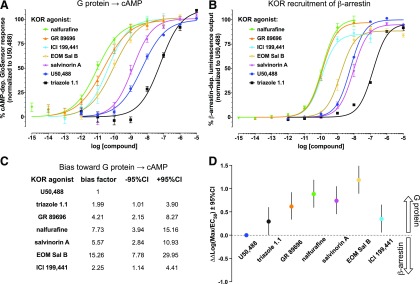
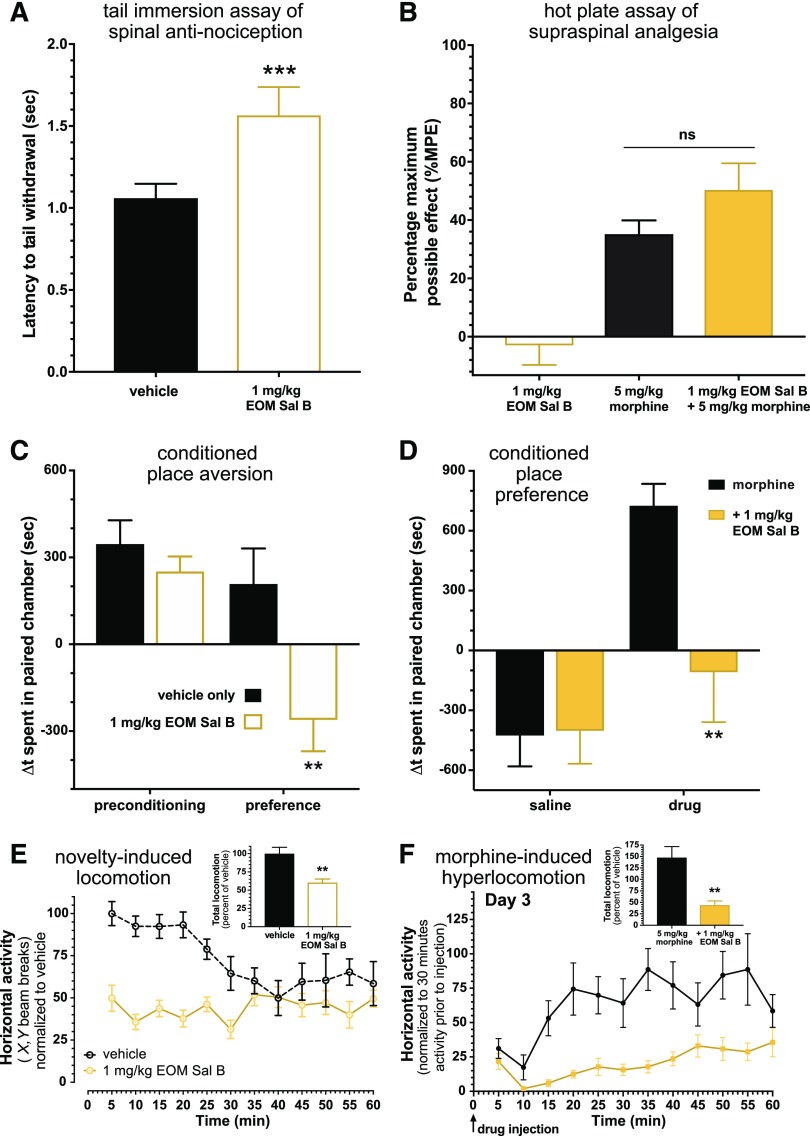
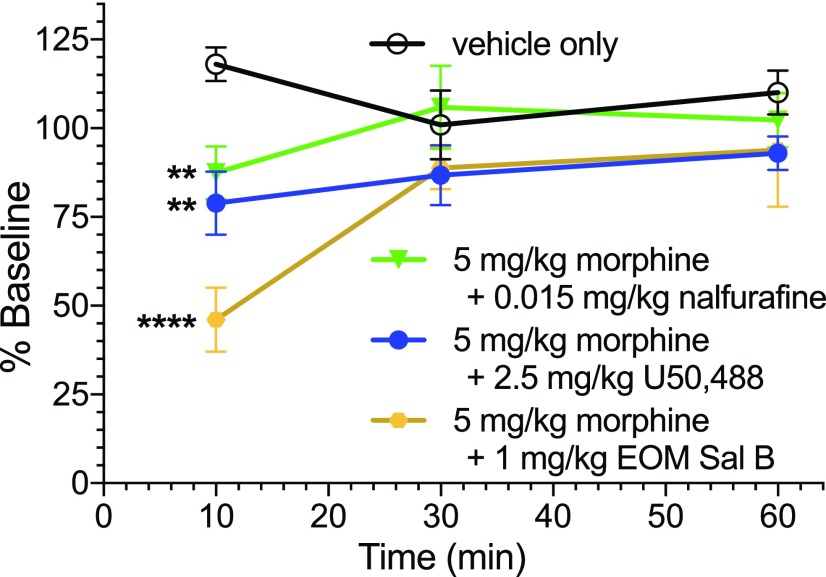
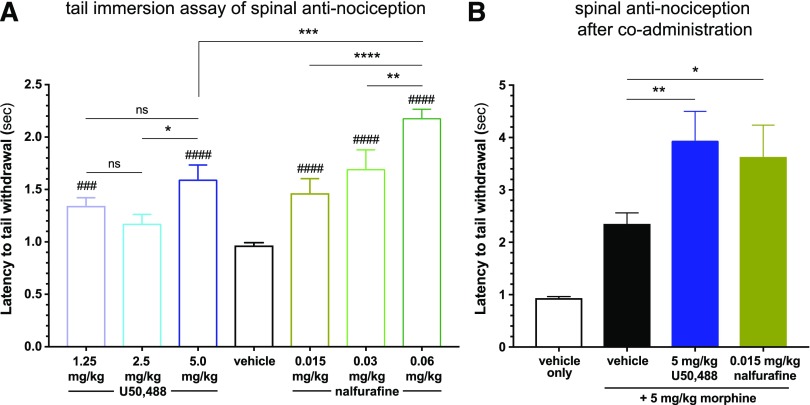
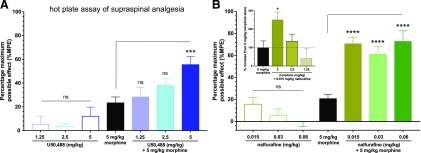
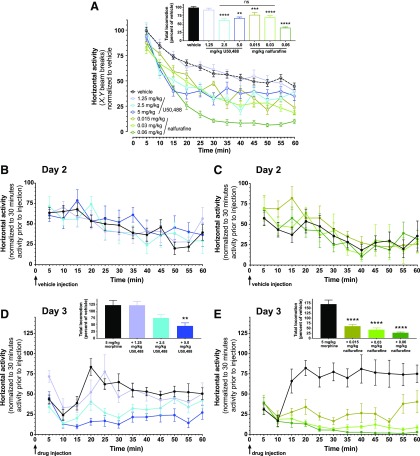
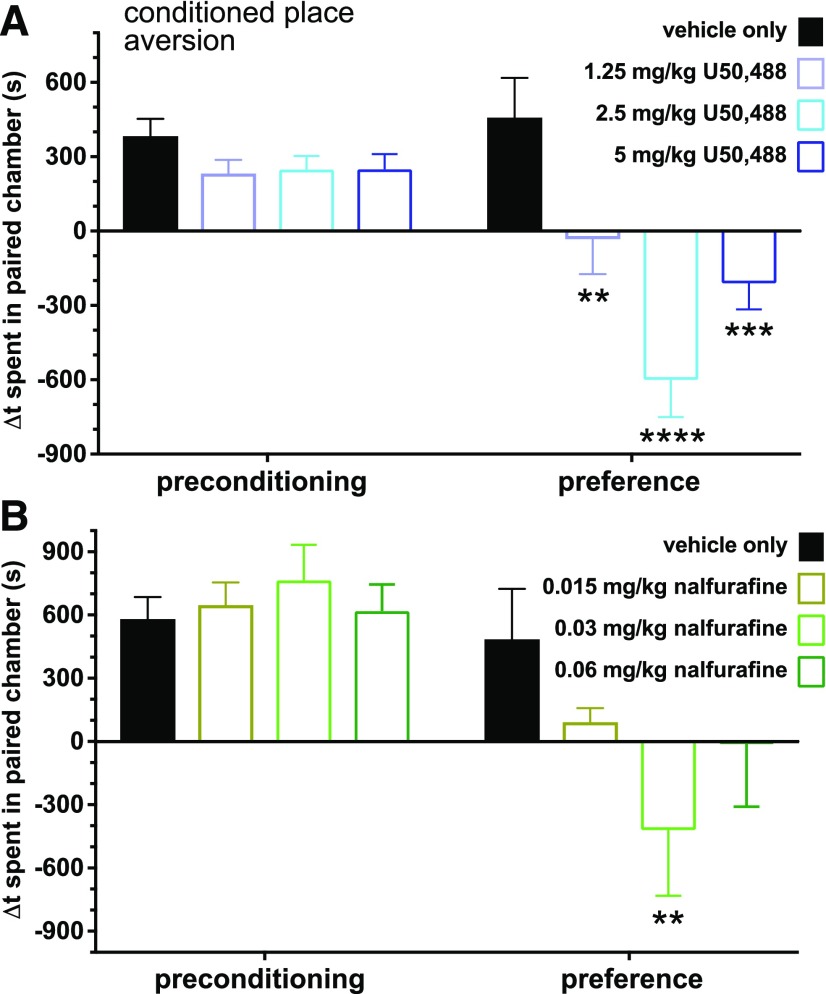
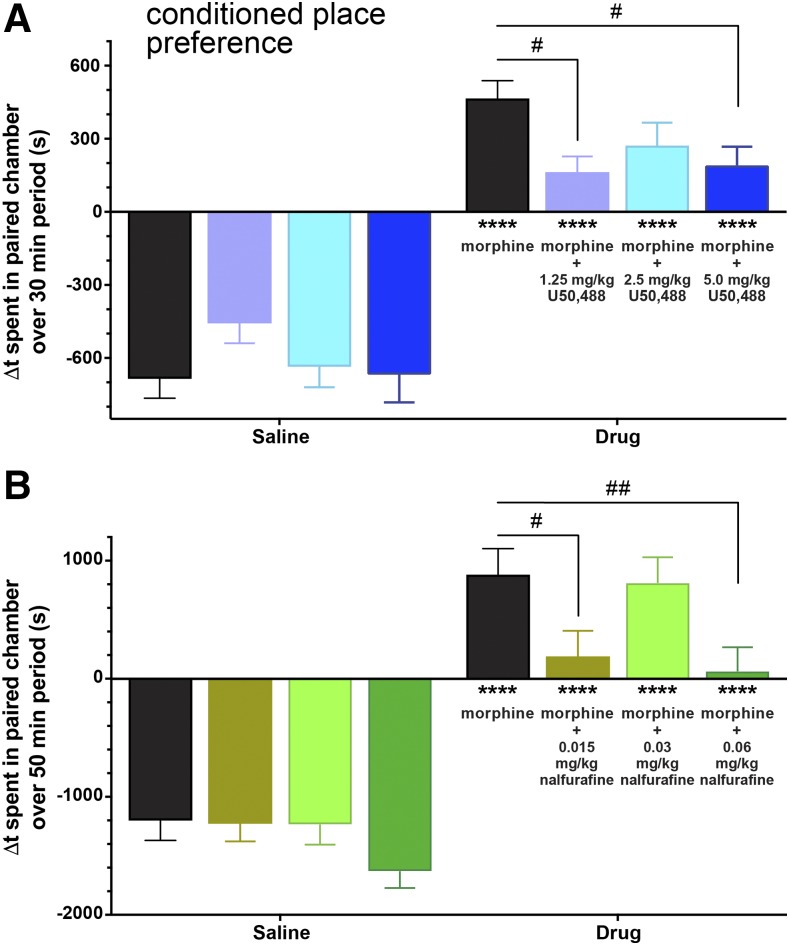
Mu opioid receptor (MOR)-targeting analgesics are efficacious pain treatments, but notorious for their abuse potential. In preclinical animal models, coadministration of traditional kappa opioid receptor (KOR)-targeting agonists with MOR-targeting analgesics can decrease reward and potentiate analgesia. However, traditional KOR-targeting agonists are well known for inducing antitherapeutic side effects (psychotomimesis, depression, anxiety, dysphoria). Recent data suggest that some functionally selective, or biased, KOR-targeting agonists might retain the therapeutic effects of KOR activation without inducing undesirable side effects. Nalfurafine, used safely in Japan since 2009 for uremic pruritus, is one such functionally selective KOR-targeting agonist. Here, we quantify the bias of nalfurafine and several other KOR agonists relative to an unbiased reference standard (U50,488) and show that nalfurafine and EOM-salvinorin-B demonstrate marked G protein-signaling bias. While nalfurafine (0.015 mg/kg) and EOM-salvinorin-B (1 mg/kg) produced spinal antinociception equivalent to 5 mg/kg U50,488, only nalfurafine significantly enhanced the supraspinal analgesic effect of 5 mg/kg morphine. In addition, 0.015 mg/kg nalfurafine did not produce significant conditioned place aversion, yet retained the ability to reduce morphine-induced conditioned place preference in C57BL/6J mice. Nalfurafine and EOM-salvinorin-B each produced robust inhibition of both spontaneous and morphine-stimulated locomotor behavior, suggesting a persistence of sedative effects when coadministered with morphine. Taken together, these findings suggest that nalfurafine produces analgesic augmentation, while also reducing opioid-induced reward with less risk of dysphoria. Thus, adjuvant administration of G protein-biased KOR agonists like nalfurafine may be beneficial in enhancing the therapeutic potential of MOR-targeting analgesics, such as morphine.
Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
Similar articles
-
Modulation of cocaine-related behaviors by low doses of the potent KOR agonist nalfurafine in male C57BL6 mice.Psychopharmacology (Berl). 2020 Aug;237(8):2405-2418. doi: 10.1007/s00213-020-05543-7. Epub 2020 May 20. Psychopharmacology (Berl). 2020. PMID: 32435819
-
Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor.Cell Signal. 2017 Apr;32:59-65. doi: 10.1016/j.cellsig.2017.01.016. Epub 2017 Jan 11. Cell Signal. 2017. PMID: 28088389 Free PMC article.
-
Effects of Intrathecal κ-Opioid Receptor Agonist on Morphine-Induced Itch and Antinociception in Mice.Reg Anesth Pain Med. 2016 Jan-Feb;41(1):69-74. doi: 10.1097/AAP.0000000000000326. Reg Anesth Pain Med. 2016. PMID: 26587674
-
Potential for Kappa-Opioid Receptor Agonists to Engineer Nonaddictive Analgesics: A Narrative Review.Anesth Analg. 2021 Feb 1;132(2):406-419. doi: 10.1213/ANE.0000000000005309. Anesth Analg. 2021. PMID: 33332902 Free PMC article. Review.
-
Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities.Molecules. 2020 Nov 2;25(21):5092. doi: 10.3390/molecules25215092. Molecules. 2020. PMID: 33147885 Free PMC article. Review.
Cited by
-
Preclinical Studies on Nalfurafine (TRK-820), a Clinically Used KOR Agonist.Handb Exp Pharmacol. 2022;271:137-162. doi: 10.1007/164_2021_443. Handb Exp Pharmacol. 2022. PMID: 33834276 Free PMC article.
-
An updated assessment of the translational promise of G-protein-biased kappa opioid receptor agonists to treat pain and other indications without debilitating adverse effects.Pharmacol Res. 2022 Mar;177:106091. doi: 10.1016/j.phrs.2022.106091. Epub 2022 Jan 29. Pharmacol Res. 2022. PMID: 35101565 Free PMC article. Review.
-
Opioid signaling and design of analgesics.Prog Mol Biol Transl Sci. 2023;195:153-176. doi: 10.1016/bs.pmbts.2022.06.017. Epub 2022 Aug 5. Prog Mol Biol Transl Sci. 2023. PMID: 36707153 Free PMC article. Review.
-
Antinociceptive Effects of Kappa-Opioid Receptor Agonists.Handb Exp Pharmacol. 2022;271:293-313. doi: 10.1007/164_2020_430. Handb Exp Pharmacol. 2022. PMID: 33387069 Review.
-
Biased Ligands at the Kappa Opioid Receptor: Fine-Tuning Receptor Pharmacology.Handb Exp Pharmacol. 2022;271:115-135. doi: 10.1007/164_2020_395. Handb Exp Pharmacol. 2022. PMID: 33140224 Review.
References
-
- Bardo MT, Bowling SL, Pierce RC. (1990) Changes in locomotion and dopamine neurotransmission following amphetamine, haloperidol, and exposure to novel environmental stimuli. Psychopharmacology (Berl) 101:338–343. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
