

Cochlear histopathology in human genetic hearing loss: State of the science and future prospects
- PMID: 31493568
- PMCID: PMC6778517
- DOI: 10.1016/j.heares.2019.107785
Cochlear histopathology in human genetic hearing loss: State of the science and future prospects
Abstract
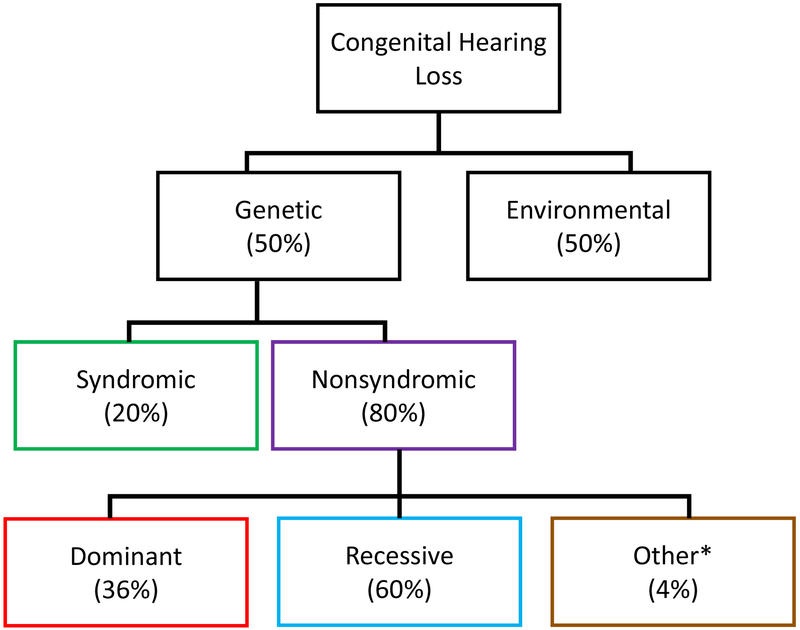
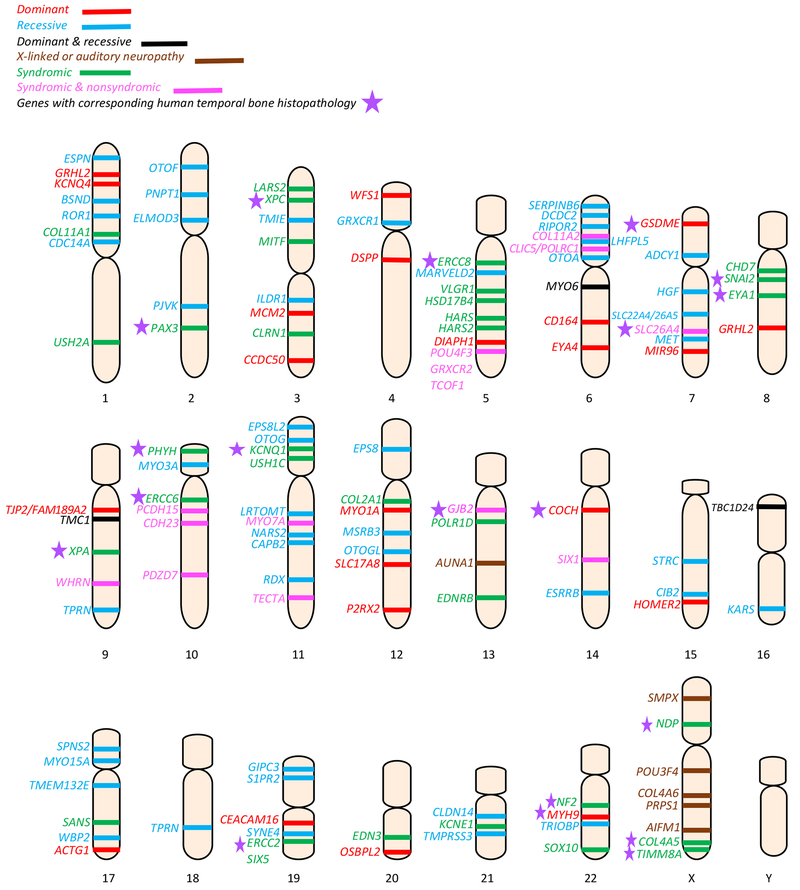
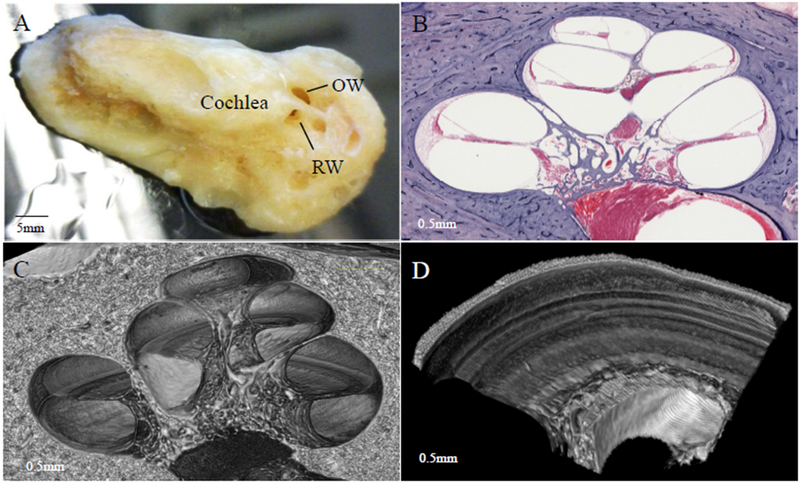
Sensorineural hearing loss (SNHL) is an extraordinarily common disability, affecting 466 million people across the globe. Half of these incidents are attributed to genetic mutations that disrupt the structure and function of the cochlea. The human cochlea's interior cannot be imaged or biopsied without damaging hearing; thus, everything known about the morphologic correlates of hereditary human deafness comes from histopathologic studies conducted in either cadaveric human temporal bone specimens or animal models of genetic deafness. The purpose of the present review is to a) summarize the findings from all published histopathologic studies conducted in human temporal bones with known SNHL-causing genetic mutations, and b) compare the reported phenotypes of human vs. mouse SNHL caused by the same genetic mutation. The fact that human temporal bone histopathologic analysis has been reported for only 22 of the nearly 200 identified deafness-causing genes suggests a great need for alternative and improved techniques for studying human hereditary deafness; in light of this, the present review concludes with a summary of promising future directions, specifically in the fields of high resolution cochlear imaging, intracochlear fluid biopsy, and gene therapy.
Keywords: Hearing loss; Histopathology; Temporal bone.
Copyright © 2019 Elsevier B.V. All rights reserved.
Figures
References
-
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP 2001. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nature Genetics 27, 99. - PubMed
-
- Avraham KB 2003. Mouse Models for Deafness: Lessons for the Human Inner Ear and Hearing Loss. Ear and Hearing 24. - PubMed
-
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA 1995. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nature Genetics 11, 369. - PubMed
-
- Bae S-H, Robertson NG, Cho H-J, Morton CC, Jung DJ, Baek J-I, Choi S-Y, Lee J, Lee K-Y, Kim U-K 2014. Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: genotype-phenotype correlations in DFNA9 deafness and vestibular disorder. Human Mutation 35, 1506–1513. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
