

Whole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han Genome
- PMID: 31494266
- PMCID: PMC6818495
- DOI: 10.1016/j.gpb.2019.07.002
Whole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han Genome
Abstract
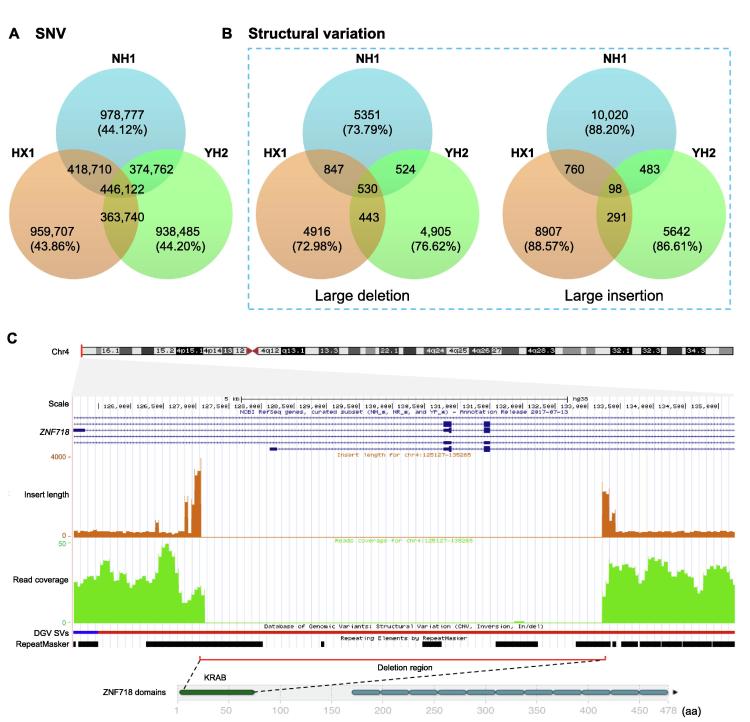
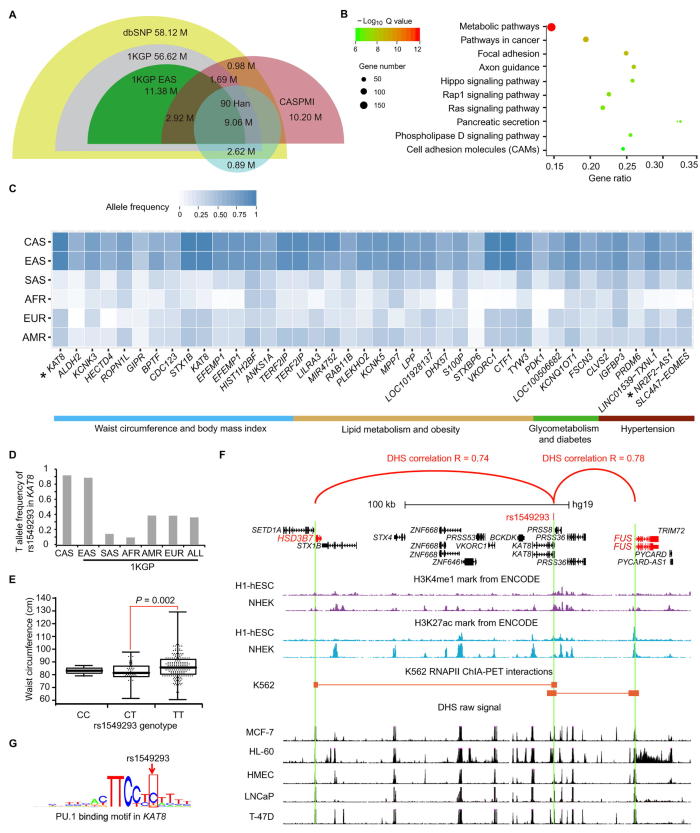
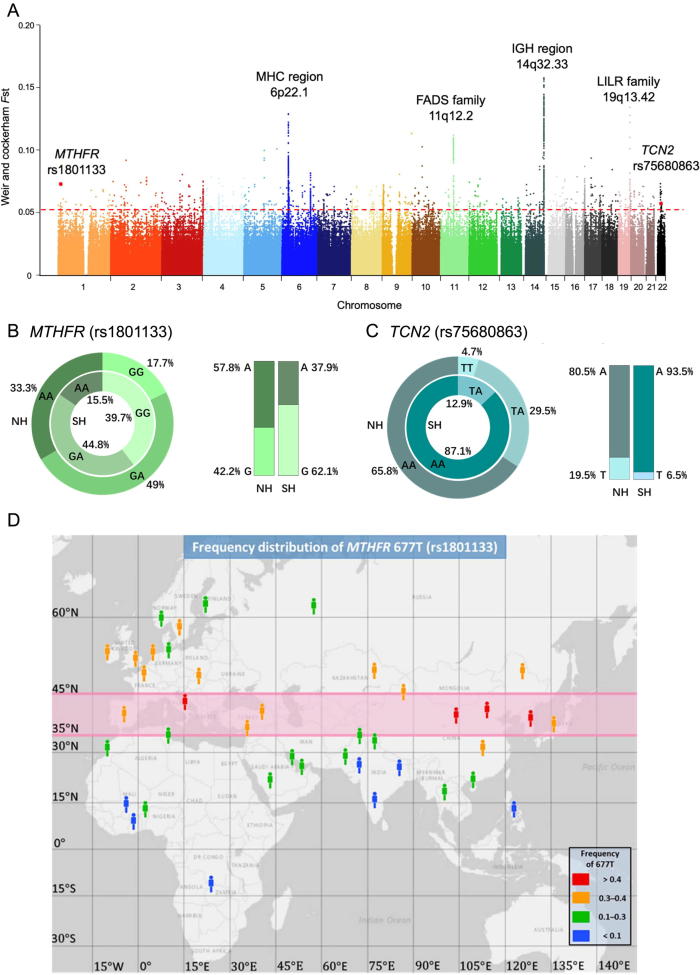
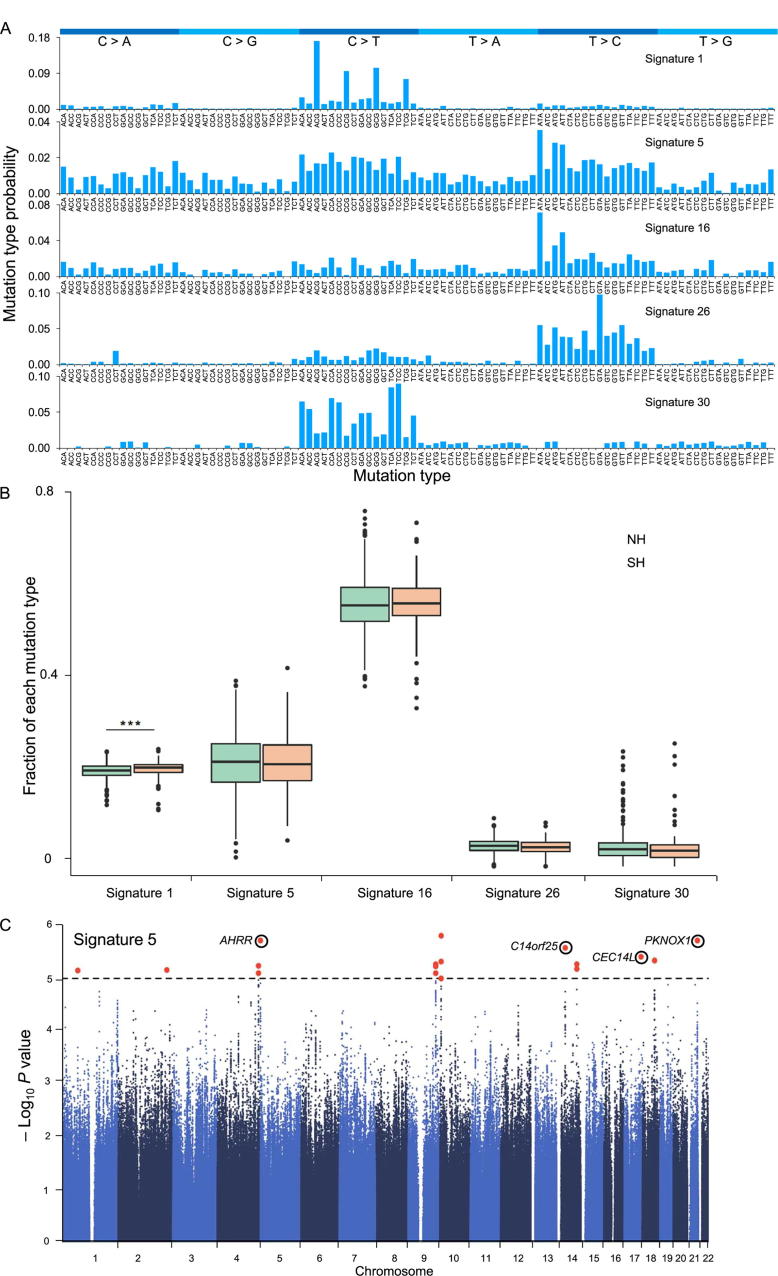
To unravel the genetic mechanisms of disease and physiological traits, it requires comprehensive sequencing analysis of large sample size in Chinese populations. Here, we report the primary results of the Chinese Academy of Sciences Precision Medicine Initiative (CASPMI) project launched by the Chinese Academy of Sciences, including the de novo assembly of a northern Han reference genome (NH1.0) and whole genome analyses of 597 healthy people coming from most areas in China. Given the two existing reference genomes for Han Chinese (YH and HX1) were both from the south, we constructed NH1.0, a new reference genome from a northern individual, by combining the sequencing strategies of PacBio, 10× Genomics, and Bionano mapping. Using this integrated approach, we obtained an N50 scaffold size of 46.63 Mb for the NH1.0 genome and performed a comparative genome analysis of NH1.0 with YH and HX1. In order to generate a genomic variation map of Chinese populations, we performed the whole-genome sequencing of 597 participants and identified 24.85 million (M) single nucleotide variants (SNVs), 3.85 M small indels, and 106,382 structural variations. In the association analysis with collected phenotypes, we found that the T allele of rs1549293 in KAT8 significantly correlated with the waist circumference in northern Han males. Moreover, significant genetic diversity in MTHFR, TCN2, FADS1, and FADS2, which associate with circulating folate, vitamin B12, or lipid metabolism, was observed between northerners and southerners. Especially, for the homocysteine-increasing allele of rs1801133 (MTHFR 677T), we hypothesize that there exists a "comfort" zone for a high frequency of 677T between latitudes of 35-45 degree North. Taken together, our results provide a high-quality northern Han reference genome and novel population-specific data sets of genetic variants for use in the personalized and precision medicine.
Keywords: De novo assembly; Large population; Phenotype association; Reference genome; Variation map.
Copyright © 2019 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
Comment in
-
Mapping Genome Variants Sheds Light on Genetic and Phenotypic Differentiation in Chinese.Genomics Proteomics Bioinformatics. 2019 Jun;17(3):226-228. doi: 10.1016/j.gpb.2019.09.001. Epub 2019 Sep 9. Genomics Proteomics Bioinformatics. 2019. PMID: 31513928 Free PMC article. No abstract available.
References
-
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Seo J.S., Rhie A., Kim J., Lee S., Sohn M.H., Kim C.U. De novo assembly and phasing of a Korean human genome. Nature. 2016;538:243–247. - PubMed
-
- Cao H., Wu H., Luo R., Huang S., Sun Y., Tong X. De novo assembly of a haplotype-resolved human genome. Nat Biotechnol. 2015;33:617–622. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
