

Platelet Extracellular Vesicles Drive Inflammasome-IL-1β-Dependent Lung Injury in Sickle Cell Disease
- PMID: 31498653
- PMCID: PMC6938158
- DOI: 10.1164/rccm.201807-1370OC
Platelet Extracellular Vesicles Drive Inflammasome-IL-1β-Dependent Lung Injury in Sickle Cell Disease
Abstract
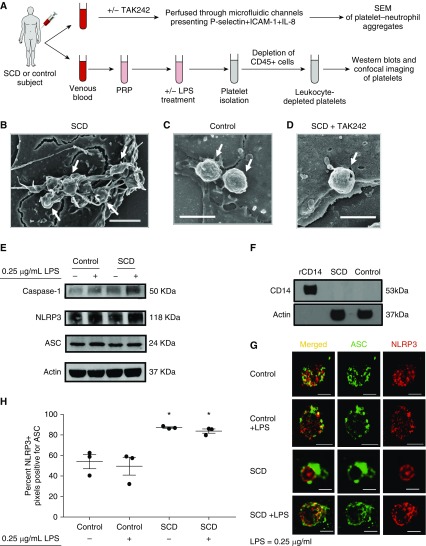
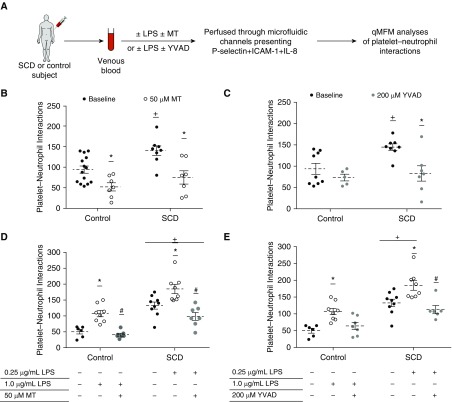
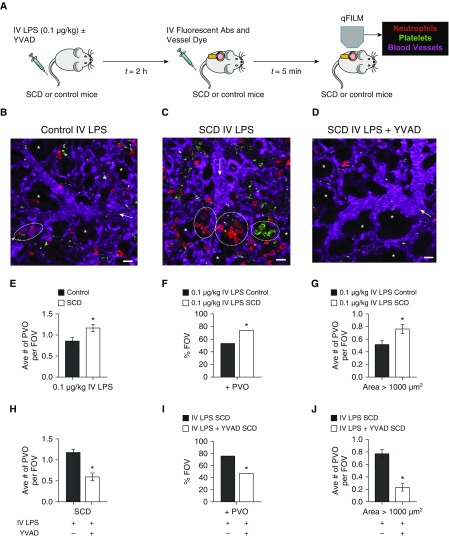
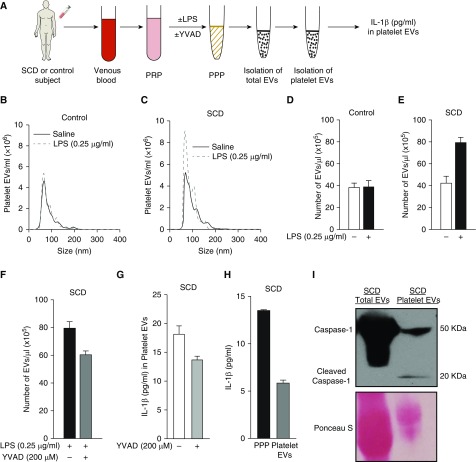
Rationale: Intraerythrocytic polymerization of Hb S promotes hemolysis and vasoocclusive events in the microvasculature of patients with sickle cell disease (SCD). Although platelet-neutrophil aggregate-dependent vasoocclusion is known to occur in the lung and contribute to acute chest syndrome, the etiological mechanisms that trigger acute chest syndrome are largely unknown.Objectives: To identify the innate immune mechanism that promotes platelet-neutrophil aggregate-dependent lung vasoocclusion and injury in SCD.Methods:In vivo imaging of the lung in transgenic humanized SCD mice and in vitro imaging of SCD patient blood flowing through a microfluidic system was performed. SCD mice were systemically challenged with nanogram quantities of LPS to trigger lung vasoocclusion.Measurements and Main Results: Platelet-inflammasome activation led to generation of IL-1β and caspase-1-carrying platelet extracellular vesicles (EVs) that bind to neutrophils and promote platelet-neutrophil aggregation in lung arterioles of SCD mice in vivo and SCD human blood in microfluidics in vitro. The inflammasome activation, platelet EV generation, and platelet-neutrophil aggregation were enhanced by the presence of LPS at a nanogram dose in SCD but not control human blood. Inhibition of the inflammasome effector caspase-1 or IL-1β pathway attenuated platelet EV generation, prevented platelet-neutrophil aggregation, and restored microvascular blood flow in lung arterioles of SCD mice in vivo and SCD human blood in microfluidics in vitro.Conclusions: These results are the first to identify that platelet-inflammasome-dependent shedding of IL-1β and caspase-1-carrying platelet EVs promote lung vasoocclusion in SCD. The current findings also highlight the therapeutic potential of targeting the platelet-inflammasome-dependent innate immune pathway to prevent acute chest syndrome.
Keywords: acute chest syndrome; neutrophil–platelet aggregates; vasoocclusion.
Figures
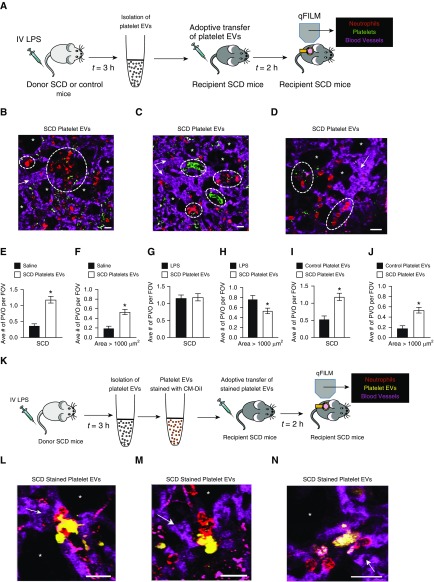
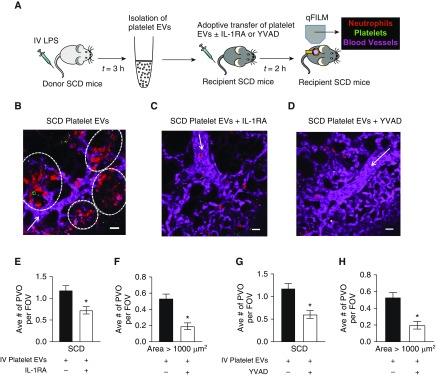
Comment in
-
Shedding New Light on Platelet Extracellular Vesicles in Sickle Cell Disease.Am J Respir Crit Care Med. 2020 Jan 1;201(1):1-2. doi: 10.1164/rccm.201909-1741ED. Am J Respir Crit Care Med. 2020. PMID: 31536373 Free PMC article. No abstract available.
References
-
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. - PubMed
-
- Chaturvedi S, Ghafuri DL, Glassberg J, Kassim AA, Rodeghier M, DeBaun MR. Rapidly progressive acute chest syndrome in individuals with sickle cell anemia: a distinct acute chest syndrome phenotype. Am J Hematol. 2016;91:1185–1190. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
