

A comparison of automatic cell identification methods for single-cell RNA sequencing data
- PMID: 31500660
- PMCID: PMC6734286
- DOI: 10.1186/s13059-019-1795-z
A comparison of automatic cell identification methods for single-cell RNA sequencing data
Abstract
Background: Single-cell transcriptomics is rapidly advancing our understanding of the cellular composition of complex tissues and organisms. A major limitation in most analysis pipelines is the reliance on manual annotations to determine cell identities, which are time-consuming and irreproducible. The exponential growth in the number of cells and samples has prompted the adaptation and development of supervised classification methods for automatic cell identification.
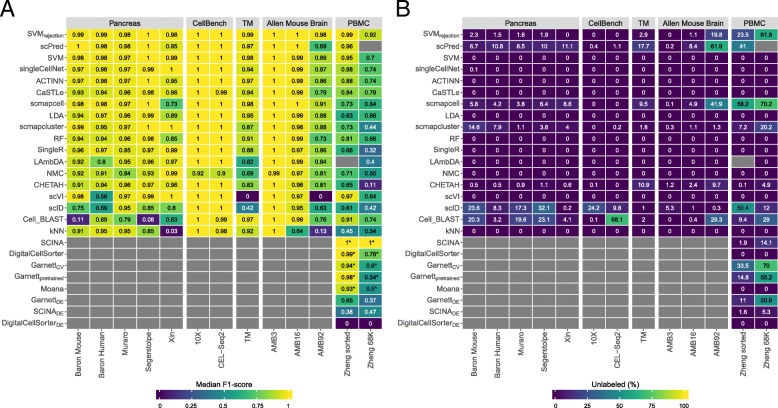
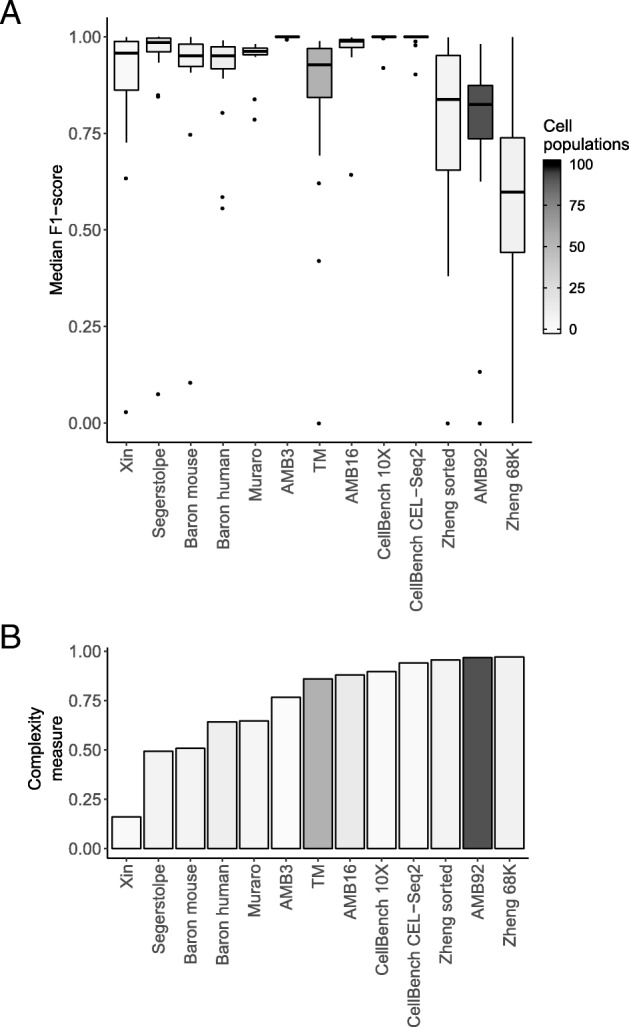
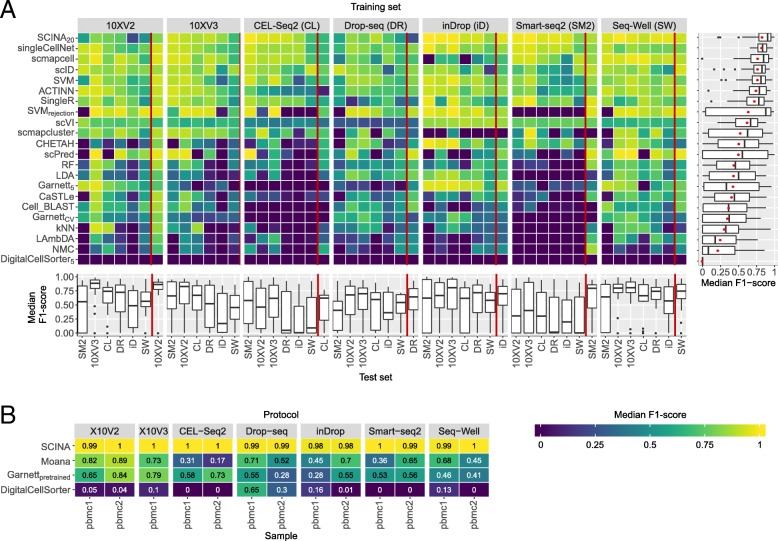
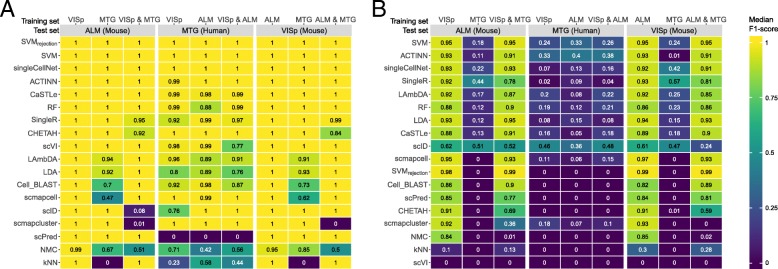
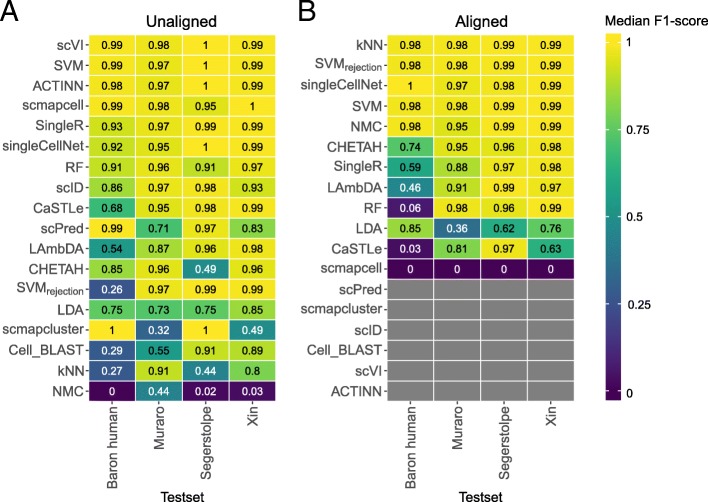
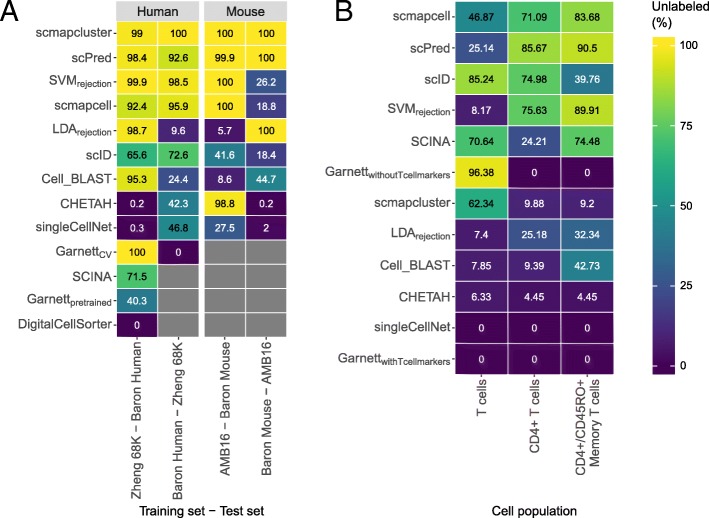
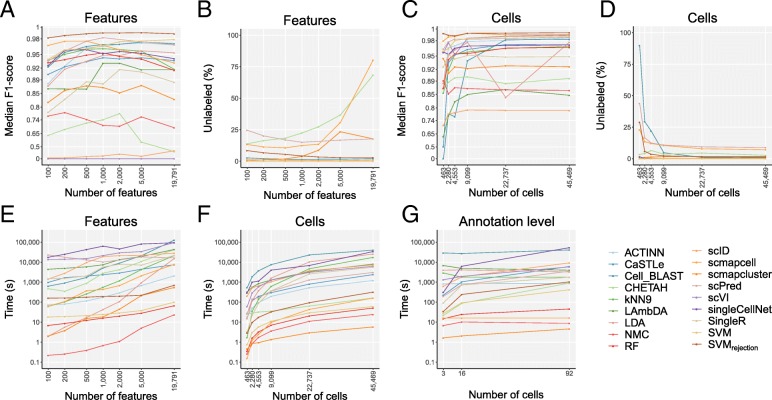
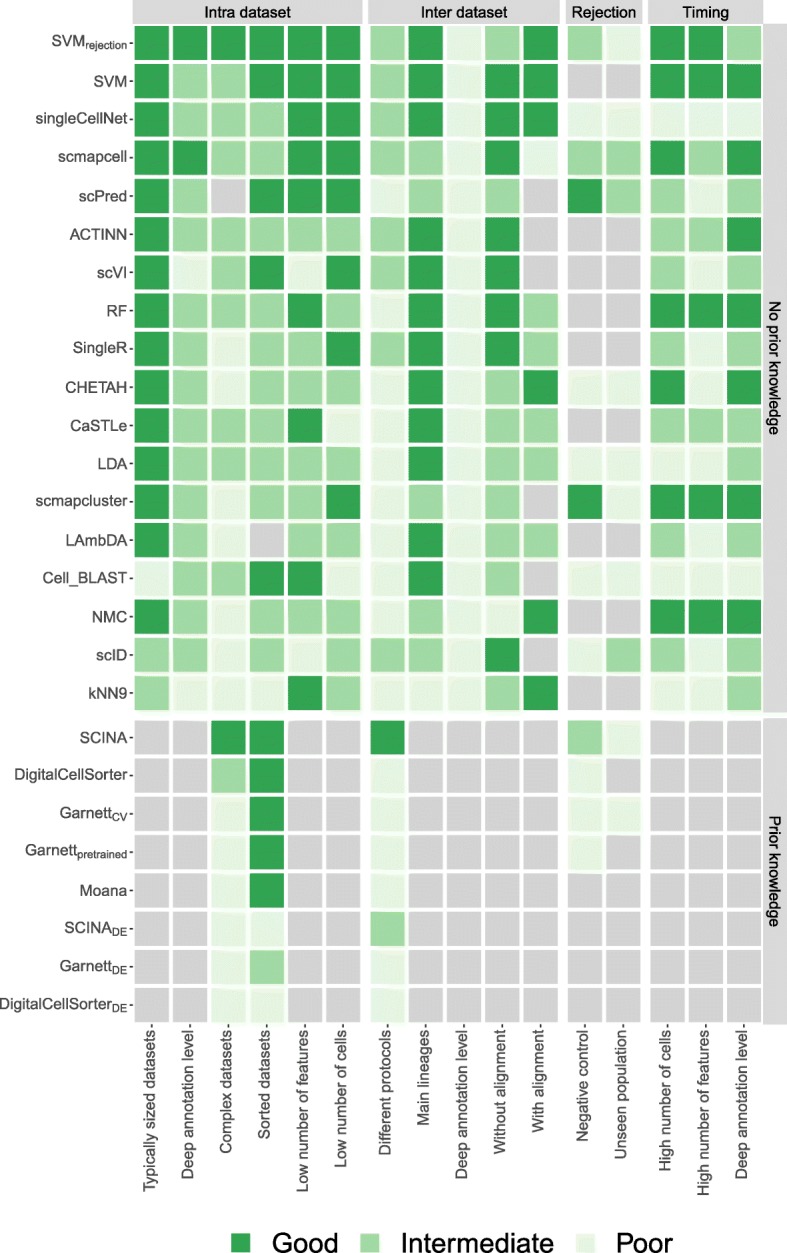
Results: Here, we benchmarked 22 classification methods that automatically assign cell identities including single-cell-specific and general-purpose classifiers. The performance of the methods is evaluated using 27 publicly available single-cell RNA sequencing datasets of different sizes, technologies, species, and levels of complexity. We use 2 experimental setups to evaluate the performance of each method for within dataset predictions (intra-dataset) and across datasets (inter-dataset) based on accuracy, percentage of unclassified cells, and computation time. We further evaluate the methods' sensitivity to the input features, number of cells per population, and their performance across different annotation levels and datasets. We find that most classifiers perform well on a variety of datasets with decreased accuracy for complex datasets with overlapping classes or deep annotations. The general-purpose support vector machine classifier has overall the best performance across the different experiments.
Conclusions: We present a comprehensive evaluation of automatic cell identification methods for single-cell RNA sequencing data. All the code used for the evaluation is available on GitHub ( https://github.com/tabdelaal/scRNAseq_Benchmark ). Additionally, we provide a Snakemake workflow to facilitate the benchmarking and to support the extension of new methods and new datasets.
Keywords: Benchmark; Cell identity; Classification; scRNA-seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Plass Mireya, Solana Jordi, Wolf F. Alexander, Ayoub Salah, Misios Aristotelis, Glažar Petar, Obermayer Benedikt, Theis Fabian J., Kocks Christine, Rajewsky Nikolaus. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science. 2018;360(6391):eaaq1723. doi: 10.1126/science.aaq1723. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
