

Proton Control of Transitions in an Amino Acid Transporter
- PMID: 31500802
- PMCID: PMC6818167
- DOI: 10.1016/j.bpj.2019.07.056
Proton Control of Transitions in an Amino Acid Transporter
Abstract
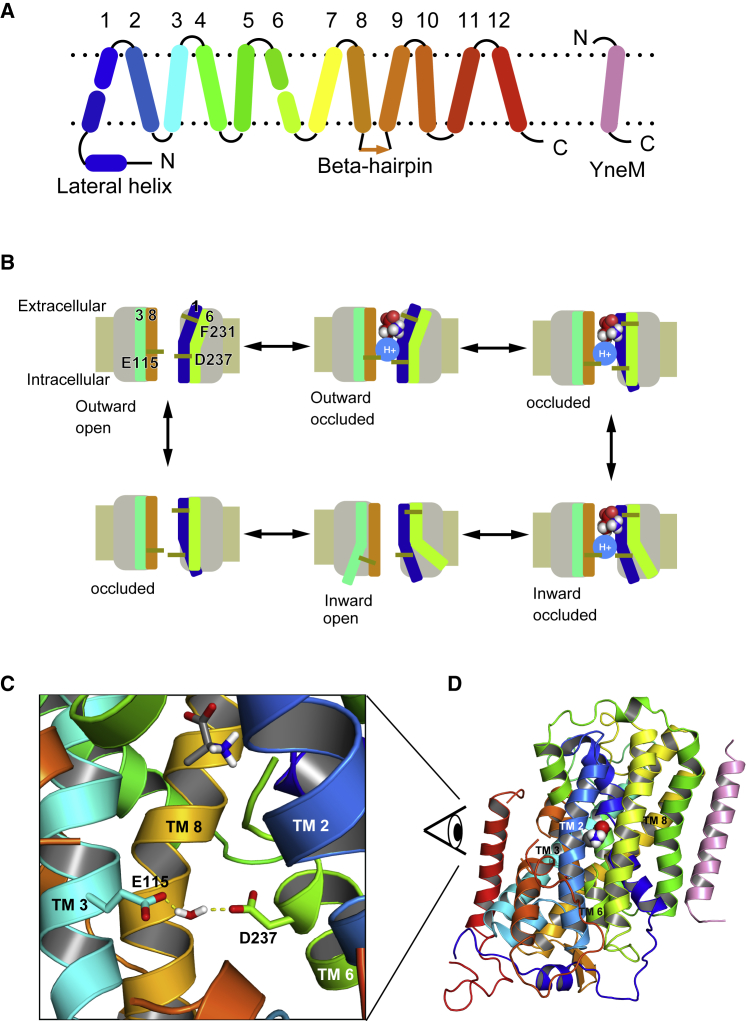
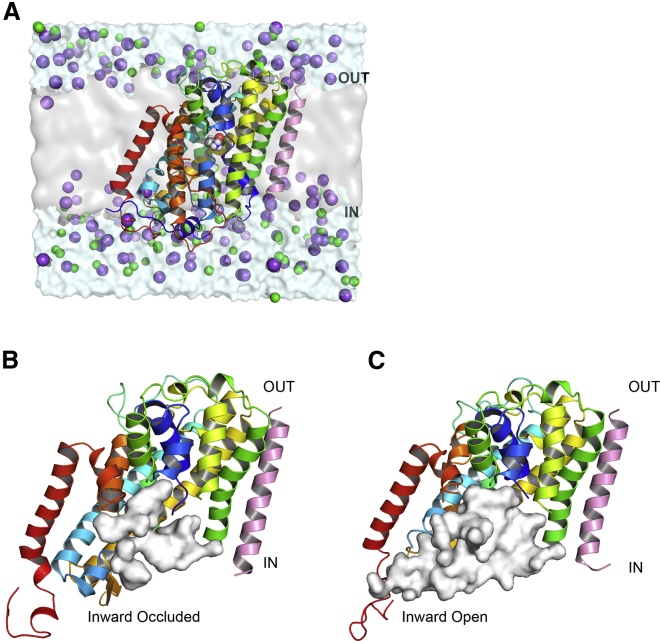
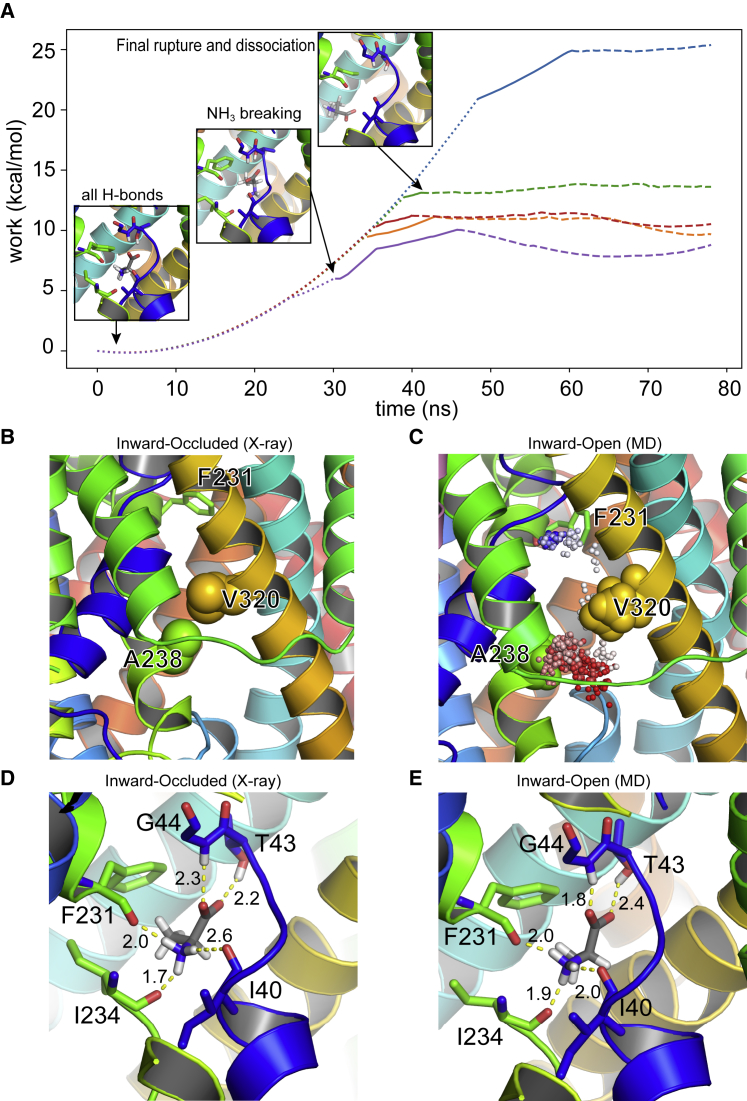
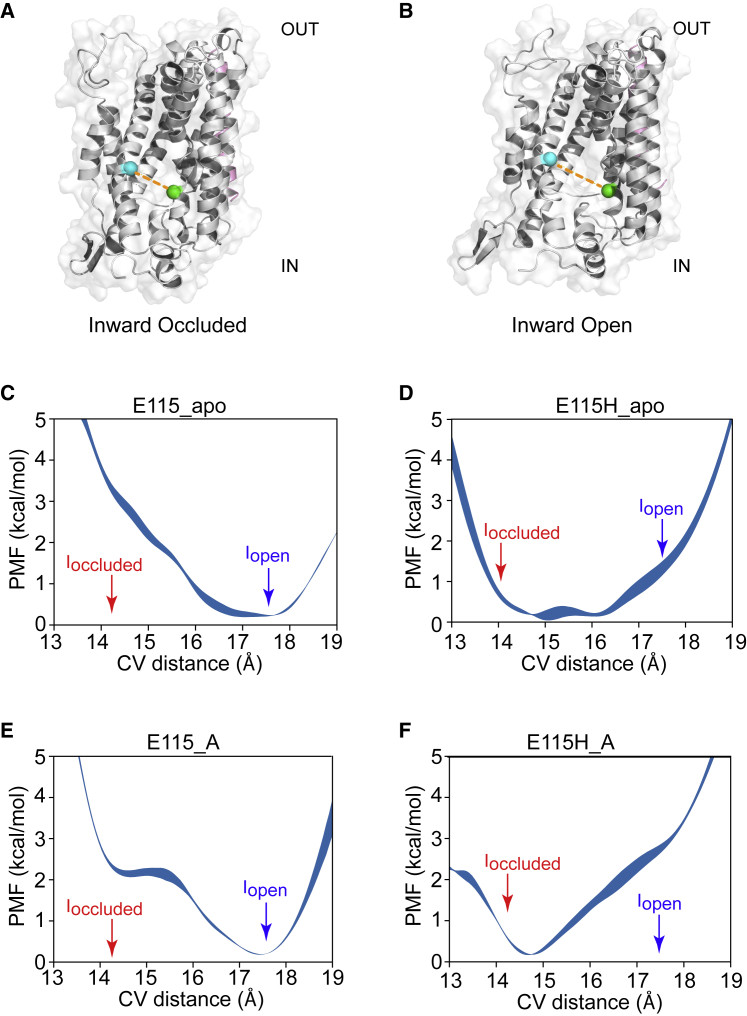
Amino acid transport into the cell is often coupled to the proton electrochemical gradient, as found in the solute carrier 36 family of proton-coupled amino acid transporters. Although no structure of a human proton-coupled amino acid transporter exists, the crystal structure of a related homolog from bacteria, GkApcT, has recently been solved in an inward-occluded state and allows an opportunity to examine how protons are coupled to amino acid transport. Our working hypothesis is that release of the amino acid substrate is facilitated by the deprotonation of a key glutamate residue (E115) located at the bottom of the binding pocket, which forms part of the intracellular gate, allowing the protein to transition from an inward-occluded to an inward-open conformation. During unbiased molecular dynamics simulations, we observed a transition from the inward-occluded state captured in the crystal structure to a much more open state, which we consider likely to be representative of the inward-open state associated with substrate release. To explore this and the role of protons in these transitions, we have used umbrella sampling to demonstrate that the transition from inward occluded to inward open is more energetically favorable when E115 is deprotonated. That E115 is likely to be protonated in the inward-occluded state and deprotonated in the inward-open state is further confirmed via the use of absolute binding free energies. Finally, we also show, via the use of absolute binding free energy calculations, that the affinity of the protein for alanine is very similar regardless of either the conformational state or the protonation of E115, presumably reflecting the fact that all the key interactions are deep within the binding cavity. Together, our results give a detailed picture of the role of protons in driving one of the major transitions in this transporter.
Copyright © 2019 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Closs E.I., Boissel J.P., Rotmann A. Structure and function of cationic amino acid transporters (CATs) J. Membr. Biol. 2006;213:67–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
