

Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes
- PMID: 31500832
- PMCID: PMC6768499
- DOI: 10.1016/j.molmet.2019.06.012
Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes
Abstract
Background: Myriad challenges to the proper folding and structural maturation of secretory pathway client proteins in the endoplasmic reticulum (ER) - a condition referred to as "ER stress" - activate intracellular signaling pathways termed the unfolded protein response (UPR).
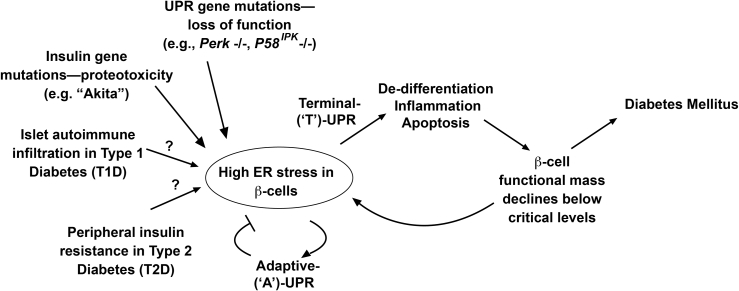
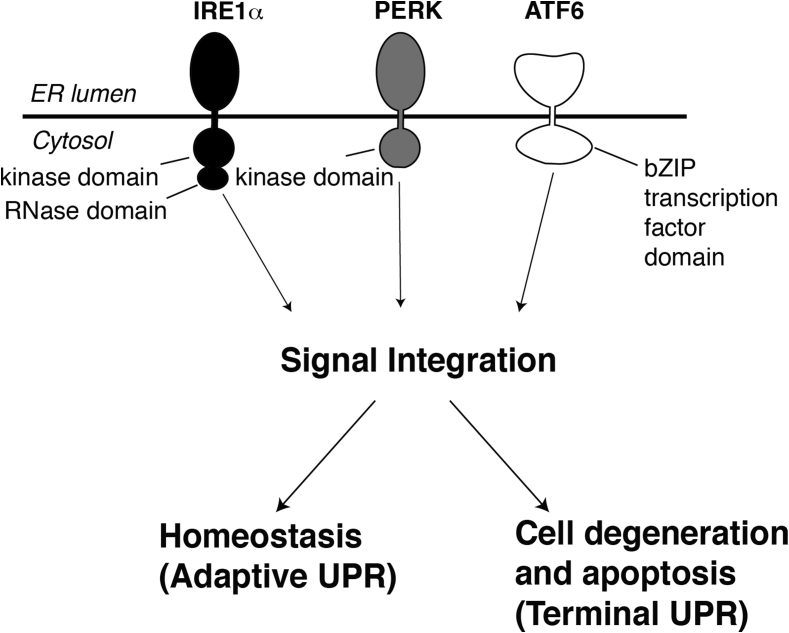
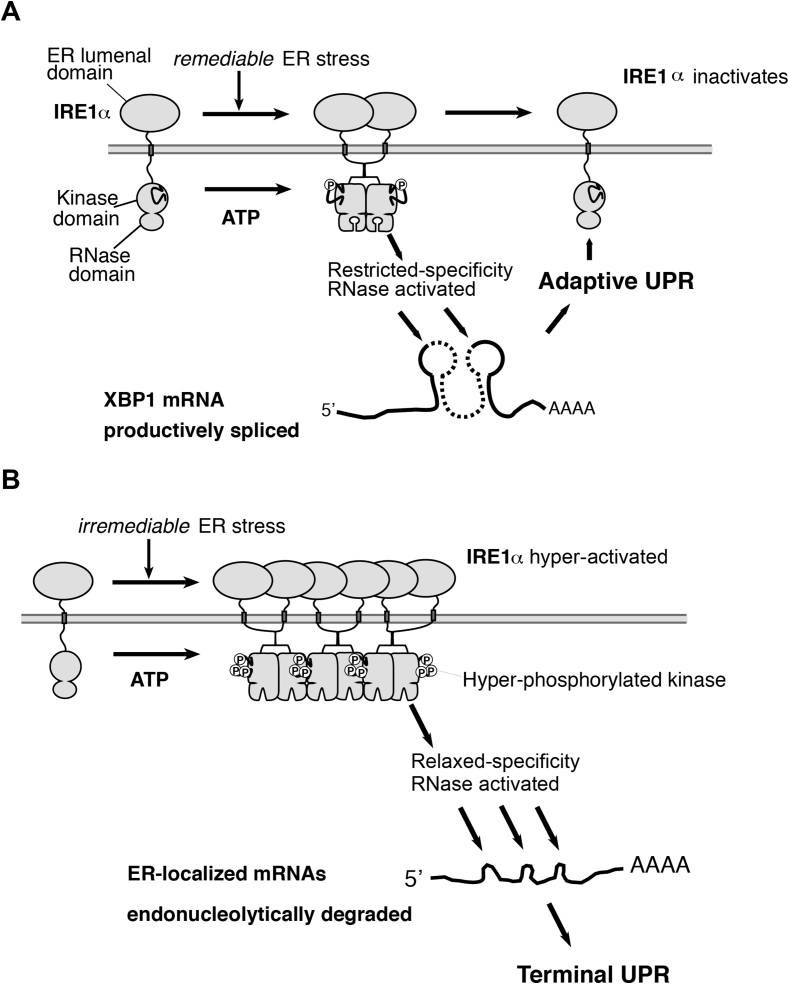
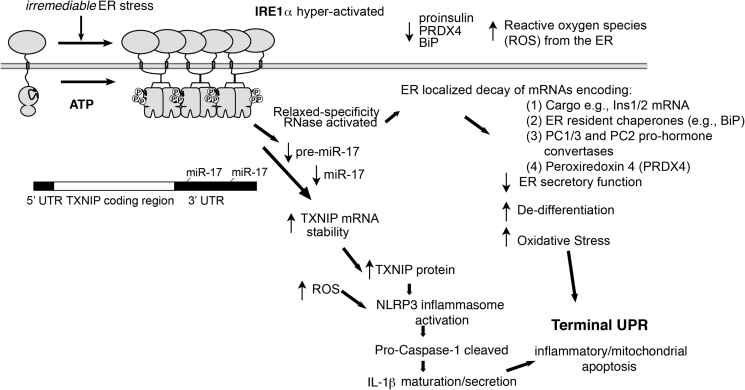
Scope of review: Through executing transcriptional and translational programs the UPR restores homeostasis in those cells experiencing manageable levels of ER stress. But the UPR also actively triggers cell degeneration and apoptosis in those cells that are encountering ER stress levels that exceed irremediable thresholds. Thus, UPR outputs are "double-edged". In pancreatic islet β-cells, numerous genetic mutations affecting the balance between these opposing UPR functions cause diabetes mellitus in both rodents and humans, amply demonstrating the principle that the UPR is critical for the proper functioning and survival of the cell.
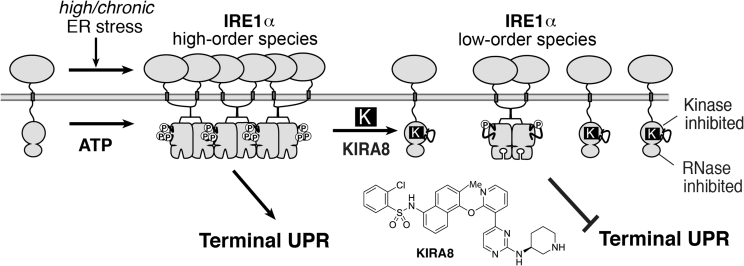
Major conclusions: Specifically, we have found that the UPR master regulator IRE1α kinase/endoribonuclease (RNase) triggers apoptosis, β-cell degeneration, and diabetes, when ER stress reaches critical levels. Based on these mechanistic findings, we find that novel small molecule compounds that inhibit IRE1α during such "terminal" UPR signaling can spare ER stressed β-cells from death, perhaps affording future opportunities to test new drug candidates for disease modification in patients suffering from diabetes.
Keywords: Apoptosis; Diabetes mellitus; Endoplasmic reticulum stress; Endoribonuclease; Kinase; Small molecule kinase inhibitor; Unfolded protein response.
Copyright © 2019. Published by Elsevier GmbH.
Figures
References
-
- van Anken E., Braakman I. Versatility of the endoplasmic reticulum protein folding factory. Critical Reviews in Biochemistry and Molecular Biology. 2005;40:191–228. - PubMed
-
- Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. - PubMed
-
- Zhang K., Kaufman R.J. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–S109. - PubMed
-
- Harding H.P., Calfon M., Urano F., Novoa I., Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annual Review of Cell and Developmental Biology. 2002;18:575–599. - PubMed
-
- Meusser B., Hirsch C., Jarosch E., Sommer T. ERAD: the long road to destruction. Nature Cell Biology. 2005;7:766–772. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
