

β-cell autophagy: Mechanism and role in β-cell dysfunction
- PMID: 31500836
- PMCID: PMC6768496
- DOI: 10.1016/j.molmet.2019.06.014
β-cell autophagy: Mechanism and role in β-cell dysfunction
Abstract
Background: Elucidation of the basic molecular mechanism of autophagy was a breakthrough in understanding various physiological events and pathogenesis of diverse diseases. In the fields of diabetes and metabolism, many cellular events associated with the development of disease or its treatment cannot be explained well without taking autophagy into account. While a grand picture of autophagy has been established, detailed aspects of autophagy, particularly that of selective autophagy responsible for homeostasis of specific organelles or metabolic intermediates, are still ambiguous and currently under intensive research.
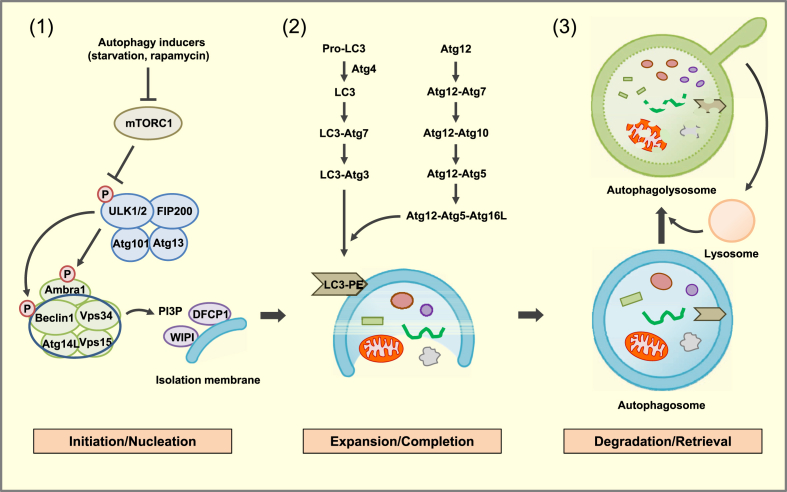
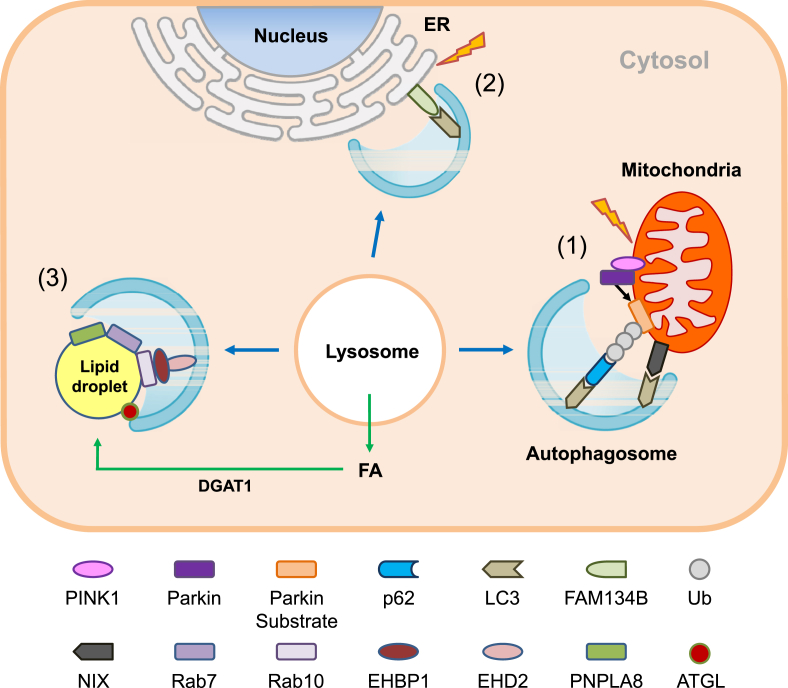
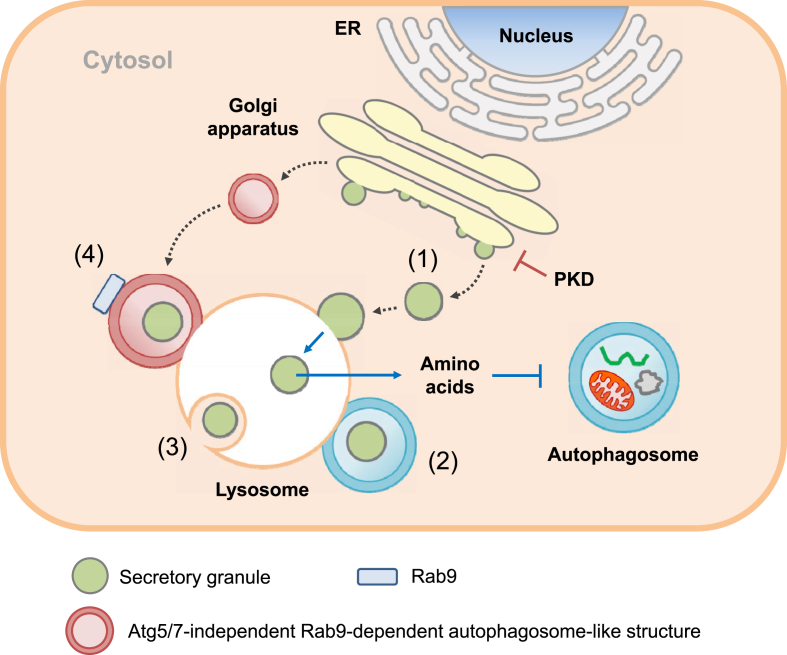
Scope of review: Here, results from previous and current studies on the role of autophagy and its dysregulation in the physiology of metabolism and pathogenesis of diabetes are summarized, with an emphasis on the pancreatic β-cell autophagy. In addition to nonselective (bulk) autophagy, machinery and significance of selective autophagy such as mitophagy of pancreatic β-cells is discussed. Novel findings regarding autophagy types other than macroautophagy are also covered, since several types of autophagy or lysosomal degradation pathways other than macroautophagy coexist in pancreatic β-cells.
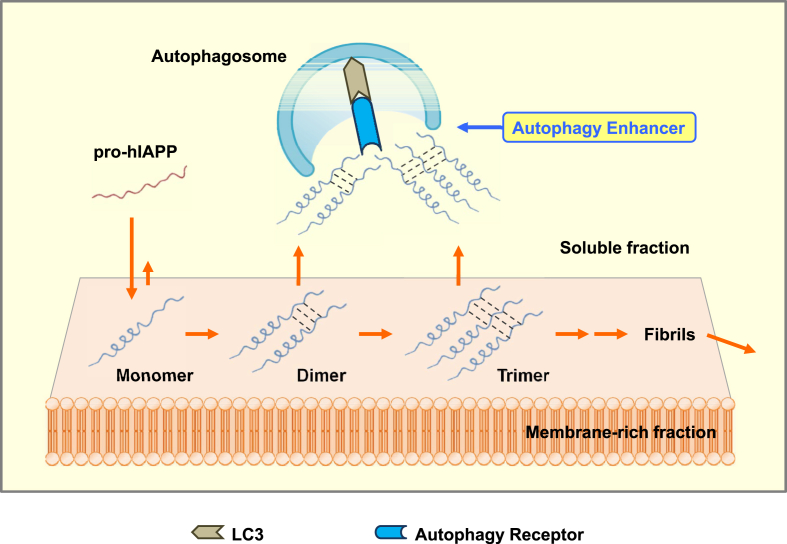
Major conclusion: Autophagy plays a critical role in cellular metabolism, homeostasis of the intracellular environment and function of organelles such as mitochondria and endoplasmic reticulum. Impaired autophagic activity due to aging, obesity or genetic predisposition could be a factor in the development of β-cell dysfunction and diabetes associated with lipid overload or human-type diabetes characterized by islet amyloid deposition. Modulation of autophagy of pancreatic β-cells is likely to be possible in the near future, which would be valuable in the treatment of diabetes associated with lipid overload or accumulation of islet amyloid.
Keywords: Autophagy; Crinophagy; Islet-associated polypeptide (IAPP); Lysosome; Mitophagy; β-cells.
Copyright © 2019. Published by Elsevier GmbH.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
